

The purpose of the Child and Family Services Act is to protect children from harm, promote the integrity of the family and assure the best interests of children. In all proceedings and matters pursuant to this Act, the paramount consideration is the best interests of the child. If this is the case then who is truly monitoring their standards, and with who’s money and interests…
Department of Justice contributes monies to Child and Family Services. In my opinion their affiliation is one that is not always designed to help children and their families, but rather one that is designed to poorly attempt to justify and defend many wrong-doings, as in this circumstance. My interpretation on “best interests” seems to be different than theirs. Their “best interests” are funded, my best interests are fundamental. Doing things from the heart and doing things from the pocket-book can have two totally different and even fatal scenarios.
 When Marco Pedersen and his wife Erica received the terrible news that no parent wants to hear at the Children’s Hospital of Eastern Ontario (CHEO), they were also faced with the damaging impact of Canada’s Child and Family Services Act, and the workers whom enforce it.
When Marco Pedersen and his wife Erica received the terrible news that no parent wants to hear at the Children’s Hospital of Eastern Ontario (CHEO), they were also faced with the damaging impact of Canada’s Child and Family Services Act, and the workers whom enforce it.
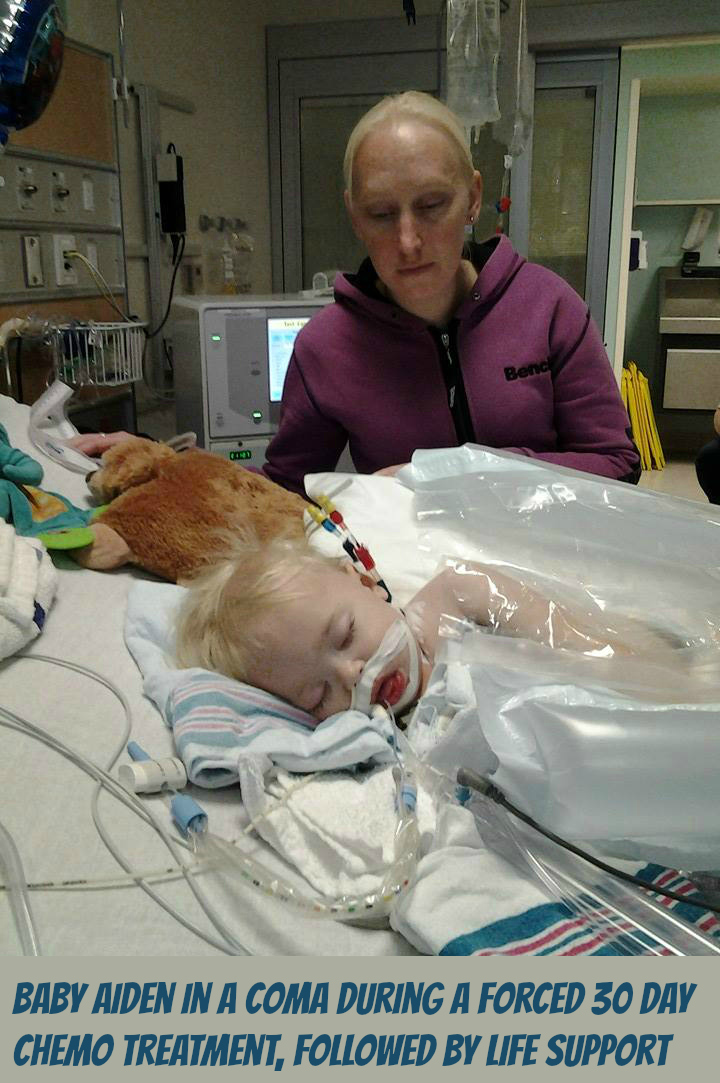
As the parents of Aiden Pedersen received the news of Aiden’s Leukemia diagnosis, Aiden’s father Marco was already aware of the dangers of Chemo and did not want Chemo administered to his child. When Aiden’s parents refused to consent for chemotherapy, the Children’s Hospital of Eastern Ontario then contacted Ottawa Children’s Aid Society. After investigation, the Children’s Aid Society went to Family Court requesting that the judge have Baby Aiden removed from his parents. The politics of government approved bullying highly pressured Marco’s wife (Erica) to be forced to consent chemotherapy for Aiden. Now, because of Marco’s beliefs he has been denied any role in Aiden’s treatment. Marco explains that they have given Aiden about 9 bags of chemo so far. “…they give him one every 2 days or so. at least thats what the first week was” “…he’s my son, so he’s a fighter, and he’s gonna beat this.” Marco then went on to say…”3.6 years of hell planned for this innocent boy” “..and they’ve yet to even release me any medical records. I’m in court monday” -Marco’s is in court Monday (Sept.29).
Eva Schacherl, a spokeswoman for CHEO, has said if one or both parents refuse a standard medical treatment and that puts their child at risk, “we’re obliged under the Child and Family Services Act to report to the CAS. The CAS is then the arbiter of determining what’s in the child’s best interests.”
“Be$t interest$”?..?

Marco said he asked the doctors at CHEO to consider alternatives to chemotherapy for Aiden, but they refused. They also rejected his request for a second opinion, and they refused to release test results and diagnosis papers.
As an alternative to chemotherapy, Marco wanted his child to have access to hemp oil as a first line of defence in Aiden’s battle against cancer. He would have preferred Aiden doing a full treatment of Cannabinoid Oil (a completely non toxic oil commonly referred to as RSO or Rick Simpson Oil) as per Rick Simpson’s Protocol versus chemo.. RSO is something Aiden’s grandfather learned about from a man who is also his neighbour, Mr. Rick Simpson himself. Rick Simpson, the Macaan man who has helped many thousands cure their own cancers and various other diseases and disorders.
Please view below the raw footage showing some of Aiden’s parents experience with Children’s Hospital of Eastern Ontario (CHEO) and a Children’s Aid Society worker. See how the parents of Aiden get treated in a time of crisis.
“Its just gonna happen” -Children’s aide worker threatening to take Marco’s son away over refusing chemo and they wouldn’t allow a second opinion or option.
…just look at the body language. The stern face and folded arms of the CHEO worker and the Children’s aide worker’s behaviour gives me the chills. Marco’s energy is focused on his son, but the CHEO and Children’s aide worker do NOT have their energy focused in the same direction.”It’s just gonna happen” says the Children’s aide worker (to me that is a hurtful, damaging & highly pressured statement of an intention to inflict pain, injury, damage, or other hostile action on someone in retribution for something done or not done (… a threat and nothing less). “I’m not gonna talk to you…I’m not gonna talk to you any further about it…I just said I’m not gonna talk any further about it.” the Children’s aide worker says. So these 2 workers bully the parents and then casually talk more amongst themselves than actually including the parents about Dr. Johnson’s role with the consent form…
…alright so I’m just gonna go…” says the Children’s aide worker as she leaves. “Okay perfect” is then heard from one of the workers… I never observed or felt any compassion among the 2 workers…I have now seen with my own eyes and felt my whole heart, the blackness that fills such CAS & CHEO workers.
Traumatic or extended separations can negatively impact children’s development, even relatively minor separations of a week or more that occur within the first two years of life are not entirely without adverse consequences for children’s development. A mother’s physical accessibility during the first years of life has important implications for supporting positive child development too.
When someone is battling cancer, especially a baby they should never have to battle cancer alone and especially not without their family…the cancer is enough trauma already. Shame on the Children’s Hospital of Eastern Ontario and the Ottawa Children’s Aid Society.
Children’s Hospital of Eastern Ontario’s Foundation Mission Statement:
“To further the physical, mental and social well-being of children and their families in eastern Ontario and western Quebec by raising, managing and dispersing funds.”

Meanwhile 75% Of Physicians in The World Refuse Chemotherapy for Themselves…

Patients get treated under a very profitable, toxic and barbaric Cancer Umbrella with primitive logic that you can “cure” cancer in a Human’s life,..BUT only if the cancer patient can survive Chemo, Radiation or even barbaric surgery,…this all has to change.
Almost everyone has some level of pain after surgery. Infection at the site of the wound is another possible problem. A lung infection (pneumonia) can occur, especially in patients with reduced lung function, such as smokers. Bleeding can happen either inside the body (internally) or outside the body (externally). Bleeding can occur if a blood vessel was not sealed off during surgery or if a wound opens up. Serious bleeding may cause the person to need another operation to find the source of the bleeding and stop it.
Blood clots can form in the deep veins of the legs after surgery, especially if a person stays in bed for a long time. Such a clot can become a serious problem if it breaks loose and travels to another part of the body, such as a lung. There is always a risk of life-threatening complications and long-term side effects…
Common side effects of chemotherapy and radiation are bone marrow suppression, sore mouth, inflamed mucous membranes, nausea and vomiting, loss of appetite, changes in taste and smell, diarrhea, dehydration, constipation, fatigue, flu-like symptoms, hair loss, skin changes, eye changes, pain, cystitis, bedwetting, pain at injection sites, inflamed vein, allergic reactions, radiation sickness, reduced bone growth, anxiety or depression, sleep problems, changes in sexuality, fluid retention, organ damage, as well as second cancers. Many of these side effects often leave cancer patients with even more prescription cocktails, which in-turn too will have their own side effects…
46:7 – What do those numbers mean to you?
I don’t know about you, but these numbers to me tell a story of government neglect, genocide and profit margins placed over the health of the Human Race.
Death By Doctor
Dr. Peter Glidden talks about the third leading cause of death in the U.S,..DEAth by Doctor (as published in The Journal of the American Association). “According to the United States Department of Health and Human Services 15,000 …*15,000 medicare patients a month are killed by M.D treatment and no one goes to jail.” “A handful of terrorists fly 2 planes into the Twin Towers, 3,500 people die and we go to war, but 15,000 people are killed by Medical Doctor treatments and we don’t bat an eyelash..” ~Dr. Peter Glidden
The tables must turn. Time to bring all those prohibitionists that are truly standing in the way of THeCure up on charges… Cannabis Prohibition is just another tool of genocide.
https://www.freedomwares.ca/governments-outlawing-cannabis-are-committing-genocide/
In Canada…
“Impeding attempt to save life 262. Every one who (a) prevents or impedes or attempts to prevent or impede any person who is attempting to save his own life, or (b) without reasonable cause prevents or impedes or attempts to prevent or impede any person who is attempting to save the life of another person, is guilty of an indictable offence and liable to imprisonment for a term not exceeding ten years.”
http://laws.justice.gc.ca/eng/acts/C-46/section-262.html
If the Child and Family Services Act’s Purpose and paramount consideration is to “protect children from harm, promote the integrity of the family and assure the best interests of children.” Then why aren’t they allowing a safer alternative to their toxic ways? Cannabis Prohibition has placed the oldest medicine known to man as the “alternative” medicine. The enforcement of Canada’s Child and Family Services Act seems to be more of a form of abuse in this case, but who is going to protect the children and their families from the abusive powers of Child and Family Services? Who?? They are already in the back pocket of the Department of Justice.
When it comes to refusing to consent to medical treatment and the provision of proper medical or other recognized remedial care or treatment that is considered essential by two duly qualified medical practitioners for the preservation of life, limb or vital organs of a child, the courts have only exemplified their abusive powers here. Chemo and Radiation cause harm. I believe that it is code to “Do No Harm”…, but it seems there has been a whole lot of harm already done.
For Dr. Robert Klaassen, a pediatric hematologist/oncologist and an “expert” in child blood diseases and cancers at CHEO, to say to the CBC News that “chemotherapy is backed up by decades of research, and that it’s important to begin treatment as soon as possible, especially with children.”. He must also be aware from his research then about the 97 percentile…
In 1994, I believe there was a 12 year study that was published looking at adults that had developed cancer (not children however…). The results were that 97% of the time Chemotherapy didn’t work.
“An important paper has been published in the Australian journal Clinical Oncology. This meta-analysis, entitled “The Contribution of Cytotoxic Chemotherapy to 5-year Survival in Adult Malignancies” set out to accurately quantify and assess the actual benefit conferred by chemotherapy in the treatment of adults with the commonest types of cancer. Although the paper has attracted some attention in Australia, the native country of the paper’s authors, it has been greeted with complete silence on this side of the world.”
“Their meticulous study was based on an analysis of the results of all the randomized, controlled clinical trials (RCTs) performed in Australia and the US that reported a statistically significant increase in 5-year survival due to the use of chemotherapy in adult malignancies. Survival data were drawn from the Australian cancer registries and the US National Cancer Institute’s Surveillance Epidemiology and End Results (SEER) registry spanning the period January 1990 until January 2004.
Wherever data were uncertain, the authors deliberately erred on the side of over-estimating the benefit of chemotherapy. Even so, the study concluded that overall, chemotherapy contributes just over 2 percent to improved survival in cancer patients. Yet despite the mounting evidence of chemotherapy’s lack of effectiveness in prolonging survival, oncologists continue to present chemotherapy as a rational and promising approach to cancer treatment.”
“Basically, the authors found that the contribution of chemotherapy to 5-year survival in adults was 2.3 percent in Australia, and 2.1 percent in the USA. They emphasize that, for reasons explained in detail in the study, these figures “should be regarded as the upper limit of effectiveness” (i.e., they are an optimistic rather than a pessimistic estimate).
Understanding Relative Risk:
How is it possible that patients are routinely offered chemotherapy when the benefits to be gained by such an approach are generally so small? In their discussion, the authors address this crucial question and cite the tendency on the part of the medical profession to present the benefits of chemotherapy in statistical terms that, while technically accurate, are seldom clearly understood by patients.
For example, oncologists frequently express the benefits of chemotherapy in terms of what is called “relative risk” rather than giving a straight assessment of the likely impact on overall survival. Relative risk is a statistical means of expressing the benefit of receiving a medical intervention in a way that, while technically accurate, has the effect of making the intervention look considerably more beneficial than it truly is. If receiving a treatment causes a patient’s risk to drop from 4 percent to 2 percent, this can be expressed as a decrease in relative risk of 50 percent. On face value that sounds good. But another, equally valid way of expressing this is to say that it offers a 2 percent reduction in absolute risk, which is less likely to convince patients to take the treatment.”
 Cannabinoids as therapeutic agents in cancer: current status and future implications.
Cannabinoids as therapeutic agents in cancer: current status and future implications.
..Cannabis sativa plant has been used for several hundreds of years both recreationally and medicinally. Centuries ago, the Chinese medicine refers to cannabis plant for pain-relief… …It contains 3 major classes of bioactive molecules; flavanoids, terpenoids and much more than 60 types of cannabinoids.. Cannabinoids exert a direct anti-proliferative effect on tumors of different origin. They have been shown to be anti-migratory and anti-invasive and inhibit MMPs which in turn degrade the extra-cellular matrix (ECM), thus affecting metastasis of cancer to the distant organs. Also, cannabinoids modulate other major processes in our body like energy metabolism, inflammation, etc. **It is important to understand which of the cannabinoid receptors are expressed and activated in different tumors as each receptor follows a different signaling mechanism.** ..Anti-cancer agents function as apoptotic, cell cycle defective or DNA damage agents. A major discovery in cancer in cannabinoid use in cancer treatment is its ability in targeted killing of tumors. Several preclinical studies suggest that ∆9-THC, other naturally occurring cannabinoids, synthetic cannabinoid agonists and endocannabinoids have anti-cancer effects in vitro against lung carcinoma, gliomas, thyroid epithelioma, lymphoma, skin carcinoma, uterine carcinoma, breast cancer, prostate carcinoma, pancreatic cancer and neuroblastoma [4]. These findings were also supported byin vivo studies and the majority of effects of cannabinoids are mediated via CB1 and CB2. The transient receptor potential vanilloid type 1 (TRPV1) has been described as an additional receptor target for several cannabinoids. In addition, the palliative effects of cannabinoids include inhibition of nausea and emesis which are associated with chemo- or radiotherapy, appetite stimulation, pain relief, mood elevation and relief from insomnia in cancer patients… ,,,…There is more focus on CB1 and CB2, the two cannabinoid receptors which are activated by most of the cannabinoids. In this review article, we will focus on a broad range of cannabinoids, their receptor dependent and receptor independent functional roles against various cancer types with respect to growth, metastasis, energy metabolism, immune environment, stemness and future perspectives in exploring new possible therapeutic opportunities. – Role of cannabinoids in regulation of cancer growth One of the important aspects of an effective anti-tumor drug is its ability to inhibit proliferation of cancer cells. Cancer cells proliferate rapidly in uncontrolled manner. Also, these cells escape death mechanism which a normal cell undergoes like apoptosis. Apoptosis is a kind of programmed cell death (PCD) mechanism which involves activation of caspase dependent and independent pathways [39]. Cannabinoids have been proved to be anti-proliferative and apoptotic drugs. This section comprises of the detailed role of cannabinoids in modulation of tumor proliferation, cell cycle and apoptosis in various cancer types. – Cannabinoids, lymphoma and leukemia cells CB1 and CB2 receptors were over-expressed in mantle cell lymphoma (MCL), and B cell non-Hodgkin lymphoma [114-115]. ∆9-THC inhibits cell viability and increased apoptosis both in vitro in EL4 and MCL cells and EL4 tumor bearing mice. In next studies the combination of ∆9-THC and other cytotoxic agents induced apoptosis in leukemia cells by MAPK/ERK pathway [114, 116]. In addition R(+)-methanandamide and WIN-55,212-2 induced apoptosis in MCL cells, was associated with ceramide accumulation and p38, depolarization of the mitochondrial membrane, and caspase activation [117]. R(+)-methanandamide also induced apoptosis in CLL cells [118]. In contrast, cannabinoids decreased cell viability as assessed by metabolic activity. The persistent expression of mammalian homolog of Atg8 with microtubule-associated protein-1 light chain-3 II (LC3 II) and p62, as well as the lack of protection from chloroquine, indicates that lysosomal degradation is not involved in this cytoplasmic vacuolation process, distinguishing from classical autophagy [119]. Paraptosis-like cell death-a third type of a programmed cell death occurred in response to cannabinoids [119]. – Role of cannabinoids in pro-metastatic mechanisms like angiogenesis, migration and invasion Migration and invasion are characteristic features of cancer cells. Carcinoma cells that are invasive have higher migratory potential which helps them to disseminate into the surrounding tissues and spread to other organs, ultimately leading to metastasis [124]. Angiogenesis, which involves growth of new vasculature has been shown to be closely related to cancer metastasis. Developing novel anti-invasive and anti-angiogenic targets would be more effective in inhibiting metastasis at earlier stage [125]. – Role of cannabinoids in cancer metastasis Met-F-AEA, WIN 55,212-2, JWH-133 and JWH-015 inhibit the migration and invasion of MDA-MB231 breast cancer cells to distant sites such as lung [27, 50, 126, 139-140]. CBD inhibits cell proliferation and invasion of 4T1 cells (mammary metastatic cell line) and reduces primary tumor volume as well as lung metastasis in 4T1-xenografted orthotopic model of nude mice [141-142]. This anti-metastatic effect was mediated by downregulation of Id-1 (a basic helix-loop-helix transcription factor inhibitor), ERK and also by inhibiting the ROS (Reactive Oxygen Species) pathway. Furthermore, CBD reduced the number of metastatic foci in 4T1- tail vein injected syngenic model. In lung cancer, JWH-015 and Win55,212-2 inhibit in vitro chemotaxis, chemoinvasion and in vivo tumor growth and lung metastasis via inhibition of AKT, matrix metalloproteinase 9 expression (MMP-9), but the pretreatment of CB1/CB2 selective antagonists, AM251 and AM630 antagonized their effects [143]. ∆9-THC inhibits growth of Lewis lung adenocarcinoma via inhibition of DNA synthesis and it suppresses growth and metastasis of A549 and SW-1573 (human lung cancer cell lines) both in vitro and in vivo due to inhibition of epidermal growth factor–induced phosphorylation of ERK1/2, c-Jun-NH2-kinase1/2 and Akt [144-145]. – Role of cannabinoids in stemness and cancer Cancer stem cells (CSC) are part of the tumor cell population. Though they might be very less in number, they have the ability to self renew and replicate to produce enormous cancer cell types. CSCs have been shown to be drug resistant with higher invasive and metastatic potential [146]. Studies show that cannabinoid receptors are involved in differentiation of neural progenitors from ectoderm and hematopoietic progenitors from mesoderm. CB1 and CB2 receptor activation modulate proliferation and differentiation of daughter progenitors. Cannabinoids- HU210, WIN55,212-2, AEA and methAEA induced concentration dependent cytotoxicity in P19 embryonal carcinoma (EC) cells [147]. It involved partial regulation by cannabinoid receptors leading to oxidative stress, necrosis coupled with apoptosis. Both CB1 and CB2 receptors are expressed in glioma stem like cells (GSC). HU-210 and JWH-133 helped in neural differentiation of GSC and blocked GSC mediated gliomagenesis [148]. These open further investigation on the function of cannabinoids and the link between stem cell and tumor progression. – Role of cannabinoids in energy metabolism and cancer One of the important by-products in energy metabolism is a set of compounds called Reactive Oxygen Species (ROS) which is produced from mitochondria and consists of H2O2, superoxide O2-, hydroxyl radical O2-, etc. Increased ROS production has been associated with triggering of apoptosis [149]. CBD modulates ERK and reactive oxygen species (ROS) pathways, which lead to down-regulation of Id-1 expression. Id-1, an inhibitor of basic helix-loop-helix transcription factors, has recently been shown to be a key regulator of the metastatic potential of breast and additional cancers [141-142]. Arachidonoyl cyclopropamide (ACPA) or GW405833 (GW) induced AMPK mediated autophagy in pancreatic adenocarcinoma cells is s
trictly related to the inhibition of energy metabolism through a ROS-dependent increase of the AMP/ATP ratio [150]. The combination of cannabinoids and gemcitabine, a nucleoside analogue used in cancer chemotherapy, synergistically inhibit pancreatic adenocarcinoma cell growth by a ROS-mediated autophagy induction without affecting normal fibroblasts [151]. Cannabidiol (CBD)-induced endoplasmic reticulum stress mediated cell death of MDA-MB231 breast cancer cells, with the coexistence of autophagy and apoptosis [63]. Recently one published report shows that ∆9-THC and ∆8-THC inhibited mitochondrial oxygen consumption rate via receptor independent manner in oral cancer cells [152]. In primary lymphocytes, treatment with CBD induced caspase 8 induced apoptosis which was mediated by oxidative stress. Similar result has been reported in glioma cells where CBD causes oxidative stress and higher enzymatic activities of glutathione reductase and glutathione peroxidase. In NSCLC cell line H460, agonists AEA, THC and HU-210 modulated the activity of mitochondrial complexes I and II-III, decreasing the mitochondrial membrane potential. ∆9-THC and cannabidiol acted synergistically to inhibit cell proliferation, modulations of the cell cycle and induction of reactive oxygen species and apoptosis as well as specific modulations of extracellular signal-regulated kinase and caspase activities in glioblastoma [107]. KM-233 induced mitochondrial depolarization, cleaved caspase 3, significant cytoskeletal contractions, and redistribution of the Golgi-endoplasmic reticulum structures in U87MG human GBM cells [109]. – Role of cannabinoids in immune environment and cancer Cancer is a type of inflammatory disease, where immune cells infiltrate into the tumor site and secrete factors which enhance the prospects of proliferation, angiogenesis and metastasis [153]. Hence, it is important to identify anti-cancer agents that target the immune related cancer environment. In glioma, WIN-55,212-2 caused accumulation of ceramide which is essential for cell death and it also had anti-inflammatory effects [154]. WIN55,212-2, abolished the PGN-activated cell growth which activates a number of inflammatory pathways, including NF-κB (aggravates tumors) and this effect was reversed by CB1 antagonist AM281 but not by the CB2 antagonist, AM630 [155]. Anandamide reduces proliferation and production of cytokines like IL-2, TNF-α and INF-γ in human T lymphocytes by activating CB2 receptor [156]. In astrocytoma and glioblastoma cells, WIN55,212-2 inhibited IL-1 mediated activation of adhesion molecules and chemokines like ICAM-1, VCAM-I and IL-8, which was receptor independent [157]. In murine T cells, cannabinol decreased IL-2 production by inhibiting nuclear factor of activated T-cells (NF-AT) and activator protein-1 (AP-1) [158]. In CD8+ T lymphocytes, JWH-133 downregulated SDF-1 induced m migration in CB2 receptor dependent manner [159].
Cannabis extract treatment for terminal acute lymphoblastic leukemia with a Philadelphia chromosome mutation.
Acute lymphoblastic leukemia (ALL) is a cancer of the white blood cells.. ..This case study is on a 14-year-old patient diagnosed with a very aggressive form of ALL (positive for the Philadelphia chromosome mutation). A standard bone marrow transplant, aggressive chemotherapy and radiation therapy were revoked, with treatment being deemed a failure after 34 months. Without any other solutions provided by conventional approaches aside from palliation, the family administered cannabinoid extracts orally to the patient. Cannabinoid resin extract is used as an effective treatment for ALL with a positive Philadelphia chromosome mutation and indications of dose-dependent disease control. The clinical observation in this study revealed a rapid dose-dependent correlation. Presentation of the Case A 14-year-old female, P.K., presented with symptoms of weakness, shortness of breath and bruising when she was taken to the Hospital for Sick Children, Toronto, Canada, on the 10th March 2006. She was diagnosed with acute lymphoblastic leukemia (ALL), with >300,000 blast cells present. Acute chemotherapy followed by a standard chemotherapy regimen went on for 6 months after the diagnosis. Upon further analysis, she was found to be positive for the Philadelphia chromosome mutation. A mutation in the Philadelphia chromosome is a much more aggressive form of ALL. When standard treatment options were unsuccessful, a bone marrow transplant was pursued. She successfully received the transplant in August 2006 and was able to be released from isolation 45 days later. She was observed posttransplant by following the presence of blast cells, noted 6 months after treatment. Consequently, in February 2007, aggressive chemotherapy procedures (AALL0031) were administered along with a tyrosine kinase inhibitor, imatinib mesylate (Gleevac), 500 mg orally twice a day. In November 2007, 9 months after the transplant, the presence of premature blast cells was observed and it was determined that another bone marrow transplant would not be effective. In February 2008, in an effort to sustain the patient, another tyrosine kinase inhibitor, disatinib (Sprycel), was administered at 78 mg twice a day with no additional rounds of chemotherapy. The patient experienced increased migraine-like headaches in June 2008. After conducting a CT scan of the head in July 2008, cerebellitis was noted. It was assumed by the primary oncologist that the blast cells could have infiltrated the CNS and be present in the brain, although none were noted in the blood. By October 2008, ten treatments of radiation therapy had been administered to the brain. On the 4th February 2009, blood was noted in the patient’s stools and a blood cell count revealed the presence of blast cells. As a result, all treatment including the disatinib was suspended and the patient’s medical staff acknowledged failure in treating her cancer. It was charted by the patient’s hematologist/oncologist that the patient ‘suffers from terminal malignant disease. She has been treated to the limits of available therapy… no further active intervention will be undertaken’. She was placed in palliative home care and told to prepare for her disease to overwhelm her body and from which she would suffer a stroke within the next 2 months.
 Cannabinoid Treatment
Cannabinoid Treatment
After this, disease progression was observed with rising counts of blast cells. The patient was receiving frequent blood transfusions and platelets during this period. Through research conducted by the patient’s family, it was observed, in a particular paper by Guzman [1] published in Nature Reviews Cancer, that cannabinoids have been shown to inhibit the growth of tumor cells in culture and in animal models by modulating key cell-signaling pathways. Cannabinoids are usually well tolerated and do not produce the generalized toxic effects of conventional chemotherapies. The family found promise in an organization known as Phoenix Tears, led by Rick Simpson who had treated several cancers with hemp oil, an extract from the cannabis plant. Rick worked with the family to help them prepare the extract.
From the 4th to the 20th of February, the patient’s blast cell count had risen from 51,490 to 194,000. The first dose of cannabinoid resin, also referred to as ‘hemp oil’, was administered orally (1 drop about the size of half a grain of rice) at 6:30 a.m. on the 21st February 2009 (day 0 in fig. 1). A 2-ounce Cannabis indica strain (known as ‘Chronic Strain’) was used to extract 7.5 ml of hemp oil using 1.2 liters of 99%-isopropyl-alcohol solution, which was boiled off in a rice cooker. Immediately after the dosing, the patient attempted to vomit; nausea had been observed previously and is common with this condition. To deal with the bitter taste and viscous nature of the hemp oil, it was suspended in honey, a known natural digestive aid, and then administered to the patient in daily doses. The objective was to quickly increase the frequency and amount of the dose and to hopefully build up the patient’s tolerance to cannabinoid resin (refer to fig. 1). The patient was observed to have periods of panic early on during administration of the hemp oil, along with increased appetite and fatigue. 
Fig. 1. Blast cell counts, days 0-15: Chronic Strain
The blast cell count reached a peak of 374,000 on the 25th February 2009 (day 5), followed by a decrease, which correlated with the increasing dose. The daily dosing is the amount administered per dose; the doses were initially given once per day up to a total of 3 times per day by day 15, and were continued with the same average frequency throughout the treatment. A decreased use of morphine for pain, an increase in euphoria symptoms, a disoriented memory and an increase in alertness were observed; these are typical with cannabinoid use.
After day 15, the original Chronic Strain had been consumed and administration of a new strain (referred to as Hemp Oil #2) was started. This was obtained by the family from an outside source. It was noted that administering the same dose yielded a decreased response in terms of the side effects of euphoria and appetite, and the patient suffered more nausea with this hemp oil. The blast cells began to increase, demonstrated in figure2. 
Fig. 2. Blast cell counts, days 18-39: Hemp Oil #2.
There is a wide amount of variance in cannabinoid concentration amongst different strains and even in the same strain with changes in growing conditions. The amount of each dose was increased to match the response of the blast cells that had been declining previously (fig. 1). After day 27, there was another peak blast cell count of 66,000 followed by a rapid decrease. There were elevated levels of urate present in the blood with corresponding joint pain; it was established that this was caused by tumor lysis syndrome of the blast cells. Allopurinol was administered.
On the 1st April 2009 (day 41), an infected central line with tunnel infection was noted on a blood test and the patient was admitted with a heavy antibiotic regimen of intravenous tazocin, gentamicin and vancomycin. On day 43, a new batch of hemp oil from an Afghan/Thai strain (referred to as Hemp Oil #3) prepared by the family was administered. A stronger psychosomatic response and increased fatigue were observed, so dosing was adjusted to 0.5 ml, shown in figure 3. Due to hospital restrictions, dosing was limited to twice a day.

Fig. 3. Blast cell counts, days 44-49: Hemp Oil #3.
A new batch of hemp oil was obtained by the family from an outside source and the dosing regimen continued twice a day, shown in figure 4.

Fig. 4. Blast cell counts, days 50-67: Hemp Oil #4.
After returning home from the hospital on the 11th April (day 51), the patient suffered from intense nausea, an inability to eat and overall weakness. On the 13th April, the patient was readmitted to the SickKids Hospital and was treated for refeeding syndrome. This was the outcome of stopping total parenteral nutrition too quickly and causing shock to the patient’s body while she was being treated for the infection. The dosing regimen was intermittent until day 59, remaining at 1-2 doses per day of 0.5 ml. As the blast cells began to increase and the patient’s appetite increased, the dosing frequency was again increased to 3 times per day starting on day 62, and the amount administered was increased from day 65.
On day 68, a new batch of medicine was obtained by the family from an outside source (referred to as Hemp Oil #5), shown in figure 5.

Fig. 5. Blast cell counts, days 69-78: Hemp Oil #5.
Dosing was maintained 3 times a day at 1.0 ml. On day 78, the patient had stomach pain in the morning and was admitted to hospital. Upon X-ray, it was noted that gastrointestinal bleeding had occurred. The patient was under a DNR order and ultimately passed away due to the bowel perforation. A prior history of pancolitis documented by CT scan in March 2009 pointed to neutropenic colitis with perforation as the cause of death. Furthermore, prior to starting on the hemp oil treatment, the patient had been extremely ill, severely underweight and had endured numerous sessions of chemotherapy and radiation therapy in the course of 34 months.
As reported by Hematology/Oncology at SickKids:
‘At admission her total WBC was 1.4, hemoglobin was 82, platelet count 8,000. She was profoundly neutropenic… a prior history of pancolitis documented by CT scan in March 2009 was neutropenic colitis with perforation… her abdomen was distended and obviously had some signs of diffuse peritonitis. The abdomen X-ray was in favour a perforation…she passed away at 10:05 in the present (sic) of family…’. Discussion Figure 6 is a summary of dose response to all the batches of hemp oil administered over a total of 78 days.

Fig. 6. Response to hemp oil treatment over 78 days.
The results shown here cannot be attributed to the phenomenon of ‘spontaneous remission’ because a dose response curve was achieved. Three factors, namely frequency of dosing, amount given (therapeutic dosing) and the potency of the cannabis strains, were critical in determining response and disease control. By viewing figure 6, it can be seen that introducing strains that were less potent, dosing at intervals >8 h and suboptimal therapeutic dosing consistently showed increases in the leukemic blast cell count. It could not be determined which cannabinoid profiles constituted a ‘potent’ cannabis strain because the resin was not analyzed. Research is needed to determine the profile and ratios of cannabinoids within the strains that exhibit antileukemic properties.
These results cannot be explained by any other therapies, as the child was under palliative care and was solely on cannabinoid treatment when the response was documented by the SickKids Hospital. The toxicology reports ruled out chemotherapeutic agents, and only showed her to be positive for THC (tetrahydrocannabinol) when she had ‘a recent massive decrease of WBC from 350,000 to 0.3′ inducing tumor lysis syndrome, as reported by the primary hematologist/oncologist at the SickKids Hospital.
This therapy has to be viewed as polytherapy, as many cannabinoids within the resinous extract have demonstrated targeted, antiproliferative, proapoptotic and antiangiogenic properties. This also needs to be explored further, as there is potential that cannabinoids might show selectivity when attacking cancer cells, thereby reducing the widespread cytotoxic effects of conventional chemotherapeutic agents. It must be noted that where our most advanced chemotherapeutic agents had failed to control the blast counts and had devastating side effects that ultimately resulted in the death of the patient, the cannabinoid therapy had no toxic side effects and only psychosomatic properties, with an increase in the patient’s vitality.
The nontoxic side effects associated with cannabis may be minimized by slowly titrating the dosing regimen upwards, building up the patient’s tolerance. The possibility of bypassing the psychoactive properties also exists, by administering nonpsychoactive cannabinoids such as cannabidiol that have demonstrated antiproliferative properties. Furthermore, future therapies could examine the possibility of upregulating a patient’s endogenous cannabinoids to help combat leukemic cells. It goes without saying that much more research and, even more importantly, phase clinical trials need to be implemented to determine the benefits of such therapies. Laboratory analysis is critical to figure out the constituents/profiles/ratios of the vast cannabis strains that show the most favored properties for exerting possible anticancer effects. Despite the nonstandardization of the medicines, the dose was readily titrated according to the biological response of the patient and produced a potentially life-saving response, namely, the drop in the leukemic blast cell count.
There has been an abundance of research exhibiting the cytotoxic effects of cannabinoids on leukemic cell lines in the form of in vitro and in vivo studies [1,2,3,4]. An oncology and hematology journal, Blood, has published numerous papers [2] over the years constructing the biochemical pathway to be elicited by the anticancer properties of cannabinoids. Our goal, upon examination of this significant case study which demonstrated complete disease control and a dose response curve, is to invest effort in and to focus on research and development to advance this therapy. An emphasis needs to be placed on determining the correct cannabinoid ratios for different types of cancer, the best method of administration, quality control and standardization of the cannabis strains and their growing conditions as well as therapeutic dosing ranges for various cancers contingent on staging and ages. Toxicity profiles favor therapies deriving from cannabis because toxicity within the body is greatly reduced and the devastating side effects of chemoradiation (i.e. secondary cancers or death) can be eliminated. It is unfortunate that this therapy does come with some unwanted psychosomatic properties; however, these might be eliminated by target therapies of nonpsychoactive cannabinoids such as cannabidiol which has garnered much attention as being a potent anti-inflammatory and possible antileukemic and anticancer agent. It is acknowledged that significant research needs to be conducted to reproduce these results and that in vitro studies cannot always be reproduced in clinical trials and the human physiological microenvironment. However, the numerous research studies and this particular clinical case are powerful enough to warrant implementing clinical trials to determine dose ranges, cannabinoid profiles and ratios, the methods of administration that produce the most efficacious therapeutic responses and the reproducibility of the results. It is tempting to speculate that, with integration of this care in a setting of full medical and laboratory support, a better outcome may indeed be achieved in the future. 
Delta9-tetrahydrocannabinol-induced apoptosis in Jurkat leukemia T cells is regulated by translocation of Bad to mitochondria.
Plant-derived cannabinoids, including Delta9-tetrahydrocannabinol (THC), induce apoptosis in leukemic cells, although the precise mechanism remains unclear. In the current study, we investigated the effect of THC on the upstream and downstream events that modulate the extracellular signal-regulated kinase (ERK) module of mitogen-activated protein kinase pathways primarily in human Jurkat leukemia T cells. The data showed that THC down-regulated Raf-1/mitogen-activated protein kinase/ERK kinase (MEK)/ERK/RSK pathway leading to translocation of Bad to mitochondria. THC also decreased the phosphorylation of Akt. However, no significant association of Bad translocation with phosphatidylinositol 3-kinase/Akt and protein kinase A signaling pathways was noted when treated cells were examined in relation to phosphorylation status of Bad by Western blot and localization of Bad to mitochondria by confocal analysis. Furthermore, THC treatment decreased the Bad phosphorylation at Ser(112) but failed to alter the level of phospho-Bad on site Ser(136) that has been reported to be associated with phosphatidylinositol 3-kinase/Akt signal pathway. Jurkat cells expressing a constitutively active MEK construct were found to be resistant to THC-mediated apoptosis and failed to exhibit decreased phospho-Bad on Ser(112) as well as Bad translocation to mitochondria. Finally, use of Bad small interfering RNA reduced the expression of Bad in Jurkat cells leading to increased resistance to THC-mediated apoptosis. Together, these data suggested that Raf-1/MEK/ERK/RSK-mediated Bad translocation played a critical role in THC-induced apoptosis in Jurkat cells.
Targeting CB2 cannabinoid receptors as a novel therapy to treat malignant lymphoblastic disease.
Introduction
..Delta-9-tetrahydrocannabinol (THC) is the major psychoactive component in marijuana.1 THC and other synthetic cannabinoids have been used as potential therapeutic agents in alleviating such complications as intraocular pressure in glaucoma, cachexia, nausea, and pain.2 Interest in the potential medicinal use of cannabinoids grew recently with the discovery of 2 cannabinoid receptors, CB1 and CB2.3 4 CB1 receptors are expressed predominantly in the brain, whereas CB2 receptors are found primarily in the cells of the immune system.1 4Furthermore, endogenous ligands for these receptors capable of mimicking the pharmacologic actions of THC have also been discovered. Such ligands were designated endocannabinoids and include anandamide and 2-arachidonoyl glycerol.5-7The physiologic function of endocannabinoids and cannabinoid receptors remains unclear.
Recently, anandamide was shown to inhibit the proliferation of human breast cancer cell lines MCF-7 and EFM-19 in vitro.8 Also, THC was shown to induce apoptosis in human prostate PC-3 cells and in C6 glioma cells in culture.9 10THC-induced apoptosis involved cannabinoid receptor–dependent8 11 or –independent pathways.9 10 Such studies have triggered interest in targeting cannabinoid receptors in vivo to induce apoptosis in transformed cells. To this end, cannabinoids were shown recently to inhibit the growth of C6 glioma cells in vivo.12 13
Cells of the immune system express high levels of CB2 receptors. It is not clear whether CB2 receptor ligation can induce apoptosis in normal or transformed immune cells. If CB2 receptor agonists can induce apoptosis in transformed immune cells, it could lead to development of a novel class of anticancer agents. In the current study, we investigated this possibility by using both murine and human leukemia and lymphoma lines as well as primary acute lymphoblastic leukemia (ALL) cells. We demonstrate that ligation of CB2 receptors can induce apoptosis in a wide range of cancers of immune-cell origin. Furthermore, we demonstrate that THC can inhibit the growth of murine lymphoma cells in vivo by inducing apoptosis and cure approximately 25% of the mice bearing the tumor. Together, the current data suggest that CB2 agonists that are devoid of psychotropic effects may constitute a novel and effective modality to treat malignancies of the immune system.
Materials and methods
Mice
Adult (6-8 weeks of age) female C57BL/6 mice were purchased from the National Institutes of Health, Bethesda, MD. The mice were housed in polyethylene cages and given rodent chow and water ad libitum. Mice were housed in rooms maintaining a temperature of 74 ± 2°F and on a 12-hour light/dark cycle.
Reagents
THC was obtained from the National Institute of Drug Abuse (Rockville, MD) and was initially dissolved in dimethyl sulfoxide (DMSO; Sigma, St Louis, MO) to a concentration of 20 mM and stored at −20°C. THC was further diluted with tissue culture medium for in vitro studies and phosphate-buffered saline (PBS) for in vivo studies. SR141716A and SR144528 were obtained from Sanofi Recherche (Montpellier, France). HU-210, anandamide, WIN55212, and JWH-015 were obtained from Tocris Cookson (Ellisville, MO).
Cell lines
The murine lymphomas (EL-4 and LSA), the murine mastocytoma (P815), the murine melanoma (B16F10), Sup-T1, a T-lymphoblastic leukemia cell line developed from an 8-year-old male, Jurkat, an acute T-lymphoblastic leukemia cell line generated from a 14-year-old male, Molt-4, an acute T-lymphoblastic leukemia cell line established from a 19-year-old male, and human glioma U251 cell line were all maintained in RPMI 1640 medium (Gibco Laboratories, Grand Island, NY) supplemented with 5% fetal calf serum (FCS), 10 mM HEPES, 1 mM glutamine, 40 μg/mL gentamicin sulfate, and 50 μM 2-mercaptoethanol. In assays examining the effect of cannabinoid agonists on tumor-cell viability and apoptosis, the concentration of FCS ranged from 0% to 5%.
Primary leukemic cells
Peripheral blood samples were obtained from 2 patients diagnosed with ALL. The samples were referred to as ALL no. 1 and ALL no. 2. ALL no. 1 was obtained from a male patient newly diagnosed with common acute lymphoblastic leukemia antigen (CALLA) (CD10)-positive non-B, non–T ALL. ALL no. 2 was obtained from a female patient newly diagnosed with terminal deoxynucleotidyl transferase (TdT)–positive T-cell ALL. Informed consent was obtained following institutional guidelines and approval was obtained from the institutional review board of Virginia Commonwealth University. Consent was provided according to the Declaration of Helsinki. The content of the lymphoblasts was greater than 70% as determined by flow cytometric analysis. Mononuclear cells were isolated by Ficoll-Paque density gradient centrifugation. In this study, the samples were cryopreserved and stored in liquid nitrogen before use. Viability after thawing was determined by trypan blue dye exclusion and was greater than 90%.
Measurement of the effect of cannabinoid receptor agonists on tumor-cell viability in vitro
Tumor cells were adjusted to 1 × 106cells/mL in medium containing 5% FCS or serum-free medium. The cells (1 × 106) were cultured in 24-well plates in 2 mL medium in the presence or absence of various concentrations of cannabinoid receptor agonists for 2 to 24 hours. Finally, the cells were harvested and washed twice in PBS, and the viable cell count was determined by trypan blue dye exclusion.
Detection of cannabinoid-induced apoptosis in vitro
Tumor cells (1 × 106 cells/well) were cultured in 24-well plates in the presence or absence of various concentrations of THC or other cannabinoid receptor agonists for 2 to 24 hours, as described above. Next, the cells were harvested, washed twice in PBS, and analyzed for the induction of apoptosis using either the terminal deoxynucleotidyl transferase–mediated end labeling (TUNEL) method or annexin V/propidium iodide (PI) method, as described elsewhere.1415 To detect apoptosis using the TUNEL method, we washed the cells twice with PBS and fixed them with 4% p-formaldehyde for 30 minutes at room temperature. The cells were next washed with PBS, permeabilized on ice for 2 minutes, and incubated with fluorescein isothiocyanate–dUTP and TdT (Boehringer Mannheim, Indianapolis, IN) for 1 hour at 37°C and 5% CO2. To detect apoptosis using the annexin V/PI method, we washed the cells twice with PBS and stained them with annexin V and PI for 20 minutes at room temperature. The cells were washed twice with PBS. The levels of apoptosis in both the TUNEL and annexin/PI assays were determined by measuring the fluorescence of the cells by flow cytometric analysis. Five thousand cells were analyzed per sample.
Measurement of tumor-cell viability and induction of apoptosis in vivo
Groups of 5 C57BL/6 mice were injected intraperitoneally (IP) with 1 × 106 EL-4 tumor cells suspended in 0.2 mL PBS. The control mice received PBS alone. Ten days later, the mice were injected with various concentrations of THC (0, 1, 3, or 5 mg/kg IP). The mice were killed 24 hours later and the EL-4 tumor cells were harvested from the peritoneal cavity by injecting 5.0 mL PBS, followed by aspiration of the peritoneal fluid from the cavity. The contaminating red blood cells were removed with red blood lysing solution (Sigma), and the tumor cells were washed twice with PBS. The number of viable cells was determined by trypan blue dye exclusion, and apoptosis was determined using the TUNEL assay. The presence of tumor cells in the peritoneal cavity was confirmed by the ability of the cells to grow in vitro and by the phenotype (Thy1+, CD4−, CD8−).
Effect of THC on survival of EL-4–challenged mice
Groups of 8 C57BL/6 mice were injected IP with 1 × 106 EL-4 tumor cells in a volume of 100 μL PBS. One day following tumor injection, the mice received daily IP injections for 14 days with 5 mg/kg THC in a volume of 500 μL PBS. Control mice received injections with the vehicle control. The mice were observed daily for signs of morbidity and were euthanized. Mice that survived for more than 60 days were rechallenged with live EL-4 cells (1 × 106) and tested for their ability to reject tumor and survive.
..Results
Expression of CB1 and CB2 receptors in EL-4, LSA, and P815 murine tumor cells
The expression of CB1 and CB2 cannabinoid receptor mRNA was determined by RT-PCR (Figure 1). This analysis revealed that all 3 murine tumor-cell lines expressed both CB1 and CB2 mRNA.
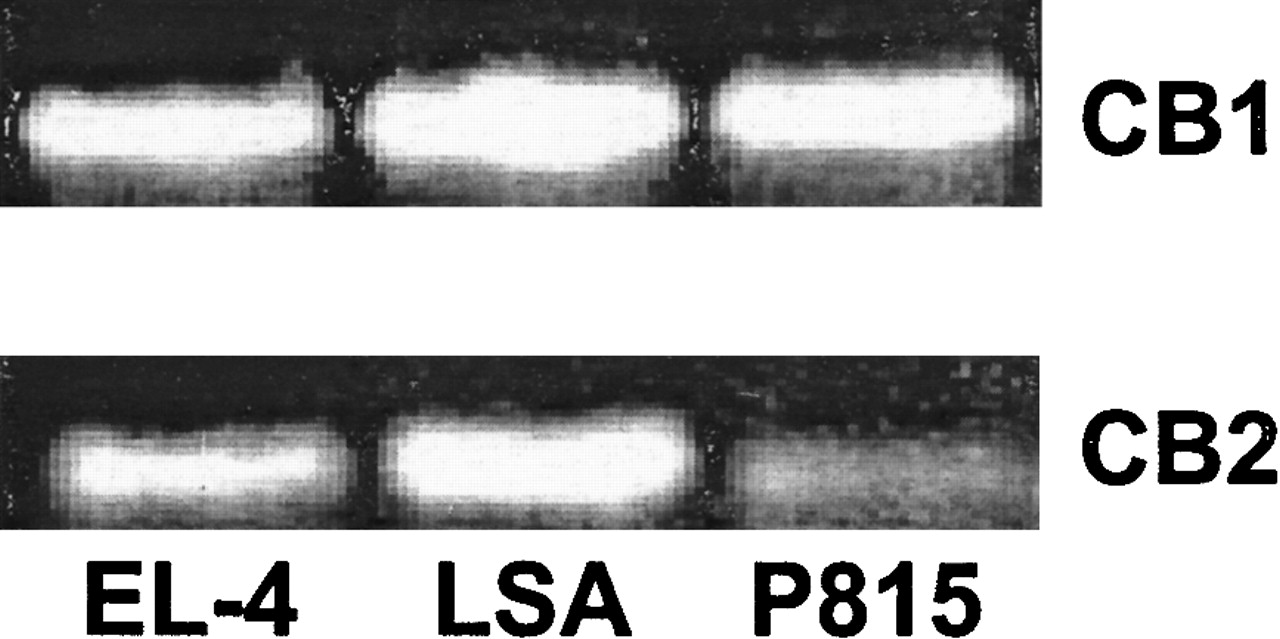 Fig. 1. The expression of CB1 and CB2 mRNA in EL-4, LSA, and P815 tumor cells.
Fig. 1. The expression of CB1 and CB2 mRNA in EL-4, LSA, and P815 tumor cells.
The expression of CB1 and CB2 mRNA was determined by RT-PCR analysis. Total RNA was isolated from EL-4, LSA, and P815 tumor cells. mRNA was reverse transcribed and amplified by PCR with primers specific for CB1 and CB2. A photograph of ethidium bromide–stained amplicons is depicted.
Exposure of EL-4, LSA, and P815 tumor cells to THC leads to a reduction in viability and induction of apoptosis in vitro
We examined whether THC exposure had an effect on the viability of EL-4, LSA, and P815 tumor cells in vitro. To this end, the tumor cells were cultured in medium containing 5% FCS and exposed to various concentrations of THC (0, 1, 10, and 20 μM) for 24 hours, and the viability was determined by trypan blue dye exclusion (Figure2A). The results showed that exposure to THC at concentrations of 10 μM or greater led to a significant reduction in the number of viable cells. Next, we analyzed the THC-treated tumor cells for induction of apoptosis by TUNEL staining (Figure 2B). The results demonstrated that THC induced significant apoptosis in all 3 cell lines in vitro. Together these results suggest that exposure of EL-4, LSA, and P815 tumor cells to THC in vitro led to significant cell killing by induction of apoptosis.

Exposure of murine tumor cells of immune origin to THC in vitro leads to a reduction in cell viability and induction of apoptosis.
(A) The effect of THC on tumor-cell viability was determined by culturing EL-4, LSA, and P815 tumor cells for 24 hours in medium containing 5% FCS in the presence of various concentrations of THC (1, 10, and 20 μM) or the vehicle. The viable cell number was determined by trypan blue dye exclusion. The data were expressed as percentage of control viable cell number. (B) The effect of THC on the induction of apoptosis in EL-4, LSA, and P815 tumor cells was determined by culturing the tumor cells for 24 hours in medium containing 5% FCS in the presence of 20 μM THC (filled histogram) or the vehicle (empty histogram). Apoptosis was quantified using the TUNEL method, and the cells were analyzed using a flow cytometer.
THC-induced effect on cellularity is dependent on exposure time and serum concentration
Previous studies suggested that the efficacy of THC may be directly related to the concentration in serum.16 17Therefore, we examined whether culturing tumor cells in serum-free medium would have an effect on THC-induced killing of tumor cells. This was accomplished by exposing EL-4 tumor cells to various concentrations of THC (1, 3, and 5 μM) or the vehicle for 4, 8, or 12 hours in serum-free medium and determining the cell viability. The results showed that by culturing the cells in serum-free medium, we dramatically reduced the concentration of THC needed to decrease tumor-cell viability (Figure 3A). For example, exposure of EL-4 tumor cells to as low as 5 μM THC for 4 hours led to a significant decrease in tumor-cell viability (Figure 3A) and an increase in the induction of apoptosis (Figure 3B,C). Also, at 12 hours, THC at a concentration of 3 μM was able to cause a significant decrease in tumor-cell viability (Figure3A). The data shown in Figure 3B and 3C demonstrate that apoptosis induced by THC was evident using both the TUNEL and annexin/PI methods. Previous studies have shown that cells positive for annexin alone represent early apoptotic cells, whereas those positive for both annexin and PI are late apoptotic/necrotic cells, and cells positive for PI alone are necrotic cells.15Thus, the majority of THC-treated cells appeared to be in an early or late apoptotic stage of death (Figure3C). It was also noted that in serum-free medium, the time required to induce tumor-cell killing was decreased significantly to 4 hours at a concentration of 5 μM THC (Figure 3A). Because serum interfered with THC-induced apoptosis, all subsequent experiments were performed in serum-free medium.

(A) EL-4 tumor cells were cultured in serum-free medium in the presence of various concentrations of THC (1, 3, and 5 μM) or the vehicle for 4, 8, or 12 hours. The number of viable cells was determined by trypan blue dye exclusion. The data represent the mean ± SEM of duplicate wells. (B) EL-4 tumor cells were cultured in serum-free medium in the presence of vehicle control (DMSO) or THC (5 μM) for 4 hours. The level of apoptosis induction was determined using the TUNEL method. (C) EL-4 cells cultured with THC as described above were stained with annexin V/PI and analyzed using a flow cytometer.
HU-210 and anandamide, but not WIN-55212, induce apoptosis in EL-4 tumor cells in vitro
Three additional cannabinoid receptor agonists were tested for their ability to induce apoptosis in EL-4 tumor cells. In Figure4A, EL-4 tumor cells were exposed to 3 μM THC, WIN55212, and HU-210 for 4 hours. The cells were then analyzed for apoptosis using the annexin/PI method (Figure 4A). The results showed that exposure to THC or HU-210 led to a significant increase in apoptosis when compared with the controls. In contrast, exposure to 3 μM WIN55212 had no significant effect on the induction of apoptosis. In addition, we examined the effects of anandamide exposure on the induction of apoptosis (Figure 4B). EL-4 tumor cells were exposed to 5 and 10 μM anandamide for 4 hours. The cells were analyzed for apoptosis using the TUNEL assay. The results showed that exposure to 5 μM led to a slight increase in apoptosis; however, exposure to 10 μM anandamide led to significant levels of apoptosis.

(A) EL-4 tumor cells were cultured in serum-free medium for 4 hours in the presence of vehicle, THC (3 μM), WIN55212 (3 μM), and HU-210 (3 μM). The level of apoptosis was quantified by annexin/PI staining, as described in Figure 3. (B) EL-4 tumor cells were cultured in serum-free medium for 4 hours in the presence of vehicle or anandamide (5 and 10 μM). The level of apoptosis was quantified by TUNEL assay.
THC treatment leads to reduced tumor burden and apoptosis in vivo
We examined whether treatment of tumor-bearing mice with THC was effective at killing tumor cells in vivo. To this end, C57BL/6 mice were injected with EL-4 tumor cells (1 × 106). On day 10 of tumor growth, the mice were injected IP with various doses of THC (1, 3, or 5 mg/kg) or the vehicle. One day later, the mice were killed and injected with 5 mL PBS into the peritoneal cavity. The peritoneal fluid was aspirated and analyzed for viable tumor cells and for apoptosis. The data demonstrated that THC caused a dose-dependent decrease in the viable tumor-cell number found in the peritoneal cavity (Figure 5A). THC failed to cause a decrease in cellularity at 1 mg/kg, but it was effective at 3 and 5 mg/kg. Furthermore, when cells collected from mice treated with 5 mg/kg were analyzed for apoptosis, a significant proportion (77.3%) of the tumor cells showed apoptosis (Figure 5B). These data suggest that THC was effective in vivo to induce apoptosis and kill the EL-4 tumor cells.
 Fig. 5.
Fig. 5.
THC treatment leads to reduced tumor burden and tumor-cell apoptosis in vivo.
C57BL/6 mice were injected IP on day 0 with 1 × 106 EL-4 tumor cells. On day 10, the mice were treated with various doses of THC (1, 3, or 5 mg/kg IP) or the vehicle. One day later, the peritoneal cavity was flushed with 5 mL PBS, and the tumor cells were collected by aspiration. (A) The cell number was determined by trypan blue dye exclusion. The data represent the mean ± SEM from groups of 3 mice. (B) The tumor cells recovered from the peritoneal cavity were tested for apoptosis using the TUNEL method. Filled histogram shows tumor cells exposed to THC and open histogram shows cells exposed to the vehicle.
THC treatment can cure tumor-bearing mice
Next we tested whether THC treatment can cure EL-4 tumor–bearing mice. To this end, mice were injected with EL-4 tumor cells (1 × 106) and then given a daily injection of 5 mg/kg THC for 14 days. The mice were observed for survival and, upon exhibiting signs of morbidity, were immediately euthanized. The results showed that treatment with THC led to a significant increase in survival (Figure 6). Interestingly, 25% of the mice survived the tumor challenge (Figure6). Also, they were completely cured inasmuch as they were resistant to rechallenge with the specific tumor (data not shown). Taken together, these results suggest that THC can exert anticancer properties in vivo.

Treatment with THC increases survival of EL-4 tumor–bearing mice.
C57BL/6 mice (8 per group) were injected IP with 1 × 106EL-4 tumor cells on day 0. From day 1 onward, the mice were treated daily for 14 days with THC (5 mg/kg) or the vehicle control by the IP route. The mice were observed daily for survival and signs of morbidity. The data depicted are representative of 3 separate experiments.
Expression of CB1 and CB2 cannabinoid receptors on human Molt-4, Jurkat, and Sup-T1 tumor-cell lines
Next, we tested whether human leukemia/lymphoma cell lines express cannabinoid receptors. The expression of CB1 and CB2 cannabinoid receptor mRNA was determined using RT-PCR analysis (Figure7). The results showed that all 3 cell lines screened expressed significant levels of CB2 mRNA. However, unlike in the murine tumor-cell lines, CB1 mRNA was not detected in these 3 cell lines. In these experiments, we used a human glioma cell line, U251, as a positive control for CB1 expression.
 Fig. 7.
Fig. 7.
The expression of CB1 and CB2 mRNA in Molt-4, Jurkat, Sup-T1, and U251 human tumor cells.
The expression of CB1 and CB2 was determined by RT-PCR analysis. Total RNA was isolated from Molt-4, Jurkat, Sup-T1, and U251 tumor cells. mRNA was reverse transcribed and amplified by PCR with primers specific for CB1 and CB2. A photograph of ethidium bromide–stained amplicons is depicted.
THC, HU-210, and anandamide induce apoptosis in human leukemia and lymphoma cell lines in vitro
Next, we examined whether exposure of human leukemia and lymphoma cell lines to THC or HU-210 would lead to induction of apoptosis. To this end, human tumor-cell lines Jurkat, Molt-4, and Sup-T1 were exposed to various concentrations of THC, HU-210 (2.5, 5, and 10 μM), or the vehicle for 4 hours, and the induction of apoptosis was determined using the TUNEL method. The results showed that exposure of the Jurkat, Molt-4, and Sup-T1 cell lines to greater than or equal to 5 μM THC or HU-210 led to significant levels of apoptosis (Figure8A). THC at 10 μM and HU-210 at 5 μM concentrations caused greater than 80% apoptosis (Figure 8A). Figure8B shows a representative experiment using the TUNEL assay. In addition, we examined the effects of anandamide exposure on the induction of apoptosis in Molt-4 tumor cells. Molt-4 tumor cells were cultured for 4 hours in the absence or presence of various concentrations of anandamide (5, 10, 20, and 40 μM). The level of apoptosis was quantified using the TUNEL method (Figure9). The results showed that anandamide at concentrations of 20 μM or greater induced significant levels of apoptosis in Molt-4 tumor cells. Together, these data suggest that THC, HU-210, and anandamide can induce apoptosis in various human leukemia and lymphoma cell lines.
 Fig. 8.
Fig. 8.
Human tumors Molt-4, Jurkat, and Sup-T1 were cultured in serum-free medium in the presence or absence of various concentrations of THC, HU-210 (2.5, 5, and 10 μM), or the vehicle for 4 hours. (A) The induction of apoptosis was determined by the TUNEL method, and the percentage of apoptotic cells was plotted. (B) A representative experiment in which human tumor cells cultured with 10 μM of THC or HU-210 (filled histograms) or the vehicle (open histograms) were analyzed for apoptosis using TUNEL assay.

Anandamide exposure leads to the induction of apoptosis in Molt-4 tumor cells in vitro.
Molt-4 tumor cells were cultured in serum-free medium in the presence or absence of various concentrations of anandamide (5, 10, 20, and 40 μM) or the vehicle for 4 hours. The induction of apoptosis was determined by the TUNEL method. A representative experiment in which Molt-4 tumor cells were cultured with anandamide (filled histogram) or the vehicle (open histogram) is depicted.
THC-induced reduction in viable cell number is mediated through the CB1 and CB2 cannabinoid receptors
Because the human tumor-cell lines screened exhibited CB2 but not CB1 receptors, we tested whether THC was acting through CB2 receptors to induce apoptosis. To this end, Jurkat and Sup-T1 cells were incubated with 5 μM THC in the presence of CB2 antagonists or the vehicle. After 4 hours, the viable cell number was determined by trypan blue dye exclusion (Figure 10). The results showed that exposure to THC led to a dramatic reduction in the number of viable tumor cells. However, when the cells were cocultured with the CB2 antagonist, the viable cell numbers increased significantly, thereby reversing the effect of THC. Together, these results suggest that THC-induced reduction in viable cell number and increase in the induction of apoptosis were mediated through the CB2 cannabinoid receptors.
 Fig. 10. CB2 receptor antagonists can reverse the toxicity of THC.
Fig. 10. CB2 receptor antagonists can reverse the toxicity of THC.
Jurkat and Sup-T1 human tumor cells were cultured for 4 hours in the presence of THC (5 μM) or the vehicle. In addition, the cultures received the CB2 antagonist (5 μM). The viable cell number was determined by trypan blue dye exclusion. The data represent the mean ± SEM of triplicate cultures.
Exposure of Jurkat and Molt-4 tumor cells to CB2 receptor agonist, JWH-015, leads to a reduction in viability and induction of apoptosis in vitro
Next we examined whether exposure to a CB2-selective agonist would lead to tumor-cell death and induction of apoptosis. Jurkat and Molt-4 tumor cells were exposed to various concentrations of the CB2-selective agonist JWH-015 (1, 5, 10, and 20 μM) or the vehicle in serum-free medium for 24 hours. The results showed that exposure of Molt-4 and Jurkat tumor cells to 5 μM or greater concentrations of JWH-015 led to a significant decrease in the number of viable tumor cells (Figure11A). Next, we examined whether exposure to JWH-015 would lead to the induction of apoptosis. Jurkat and Molt-4 tumor cells were exposed to JWH-015 for 24 hours, and apoptosis was determined by TUNEL assay (Figure 11B). The results showed that exposure of Jurkat and Molt-4 tumor cells to 5 μM JWH-015 led to significant induction of apoptosis. Together these results suggest that treatment of Jurkat and Molt-4 tumor cells with CB2-selective agonist can lead to a significant reduction in cell viability and induction of apoptosis.
 Fig. 11.
Fig. 11.Exposure to the CB2-selective agonist JWH-015 leads to reduced cell viability and induction of apoptosis in Jurkat and Molt-4 tumor cells in vitro.
(A) The effect of JWH-015 on tumor-cell viability was determined by culturing Jurkat and Molt-4 tumor cells for 24 hours in serum-free medium in the presence of various concentrations of JWH-015 (1, 5, 10, and 20 μM) or the vehicle. The viable cell number was determined by trypan blue dye exclusion. (B) The effect of JWH-015 on the induction of apoptosis in Jurkat and Molt-4 tumor cells was determined by culturing the tumor cells for 24 hours in serum-free medium in the presence of 5 μM JWH-015 (filled histogram) or the vehicle (empty histogram). Apoptosis was quantified using the TUNEL method, and the cells were analyzed using a flow cytometer.
THC induces apoptosis in primary ALL cells in vitro
Next we examined whether exposure of primary ALL cells to THC would have any effect on tumor-cell viability or induction of apoptosis. To this end, lymphoblasts isolated from peripheral blood of 2 patients with ALL (ALL no. 1 and ALL no. 2) were cultured in the presence of various concentrations of THC (1, 5, and 10 μM) or vehicle (DMSO) for 2 hours. The viable cellularity was determined by trypan blue dye exclusion. The results showed that exposure of the ALL samples to 5 μM or greater concentrations of THC resulted in significant reduction in viability (Figure12A). In addition, we examined the effect of THC exposure on the induction of apoptosis using the TUNEL method and observed that exposure of cells from both ALL patients to 5 μM or greater concentrations of THC led to significant induction of apoptosis (Figure 12B). These results were further corroborated by staining the ALL cells with annexin V and PI (data not shown). Together, these results suggest that exposure of primary ALL cells to THC can lead to significant tumor killing mediated by the induction of apoptosis.

Discussion
In the current study, we demonstrated that THC and other cannabinoids can induce apoptosis in murine and human leukemia and lymphoma cell lines as well as primary ALL cells. The human tumor-cell lines screened expressed CB2 but not CB1 receptors, whereas the murine tumors expressed both CB1 and CB2 receptors. Ligation of CB2 receptors was sufficient to induce apoptosis inasmuch as CB2-selective agonists could induce apoptosis in tumor cells. Also, THC-induced apoptosis in human tumor-cell lines was reversed by CB2 antagonists. THC was effective not only in vitro but also in vivo, as demonstrated by its ability to induce apoptosis and decrease the tumor load. Moreover, THC treatment could cure approximately 25% of the mice bearing a syngeneic tumor. Together, our data suggest for the first time that targeting CB2 receptors on tumor cells of immune origin may constitute a novel approach to treating such cancers.
The interactions between cannabinoids and their receptors in regulating neurobehavioral functions have been extensively studied. Cannabinoids have also been shown to alter immune functions, although the precise mechanisms remain unclear. Also, the physiologic functions of cannabinoid receptors on immune cells and the role played by endocannabinoids in immune-cell regulation remain unresolved. Recent studies from our laboratory demonstrated that administration of THC into C57BL/6 mice led to a marked decrease in the cellularity of the thymus and spleen that resulted from the induction of apoptosis in immune cells (manuscript submitted for publication). Recently, cannabinoids were also shown to induce apoptosis in tumor cells in vitro.9 1012 18 19 Together, such studies suggest the possible use of cannabinoids as anticancer agents.
The exact mechanism by which THC induces apoptosis in normal and transformed lymphocytes remains unclear. It is believed that THC and other cannabinoids can act by 2 distinct mechanisms. Because of its lipophilic properties, it was thought that THC acted through direct intercalation into the cell membrane. However, it was soon realized that the activity of cannabinoids was highly stereospecific, suggesting that the lipophilic properties were not solely responsible for the cannabinoids’ activity. Since then, receptors for cannabinoids have been characterized. These receptors share only 44% homology, but most cannabinoids tested show similar binding affinity to both receptors.20 Both receptors are coupled to G-protein, suggesting that endogenous cannabinoids may play a role in cell signaling.1 Therefore, it is possible that the observed effects of THC on the immune response, including the induction of apoptosis, may be mediated by signals initiated through these receptors. For example, Galve-Roperh et al12 demonstrated that apoptosis induced by THC in C6 glioma cells in vivo involved a cannabinoid receptor–dependent pathway. In contrast, others have shown in C6 glioma or a prostate cancer cell model that THC-induced apoptosis was independent of the involvement of the CB1 and CB2 receptors.
In the current study, several observations suggested that ligation of the CB2 receptor can induce apoptosis in tumors of immune origin. For example, the human tumor cells such as Jurkat and Sup-T1 expressed only CB2 receptors, and the THC-induced apoptosis in these tumor cells was inhibited at least in part by CB2 antagonists. These studies, however, did not rule out the possibility that ligation of CB1 receptors on murine tumors of immune origin would also induce apoptosis. In fact, we have observed that addition of CB1 antagonist to the EL-4 tumor cells can also inhibit the apoptosis induced by THC (R.J.M., P.S.N., M.N., unpublished results, August 2001). It should be noted, however, that the cannabinoid receptor antagonists can act as inverse agonists21 22 and thereby prevent apoptosis through an alternate pathway. It is for this reason that we used in the current study human cell lines that expressed only the CB2 receptors and showed using CB2 antagonists that ligation of CB2 receptors alone is sufficient to induce apoptosis.
THC is well known for its impact on the cytokine network.23 For example, the presence of THC or activation of the CB1/CB2 receptors can block forskolin-induced accumulation of cyclic adenosine monophosphate (cAMP),24-26 and reduced cAMP levels correlate with the repression of interleukin-2 (IL-2) transcription and secretion.27 IL-2 plays an important role in the regulation of apoptosis.28-30 Therefore, reduction in the levels of IL-2 or other cytokines following exposure to THC may partly account for increased apoptosis. In fact, previous studies from our laboratory have demonstrated that IL-2 can act as an autocrine growth factor in the autonomous proliferation of transformed T cells.31 32 Thus, inhibition of IL-2 production by THC could lead to decreased proliferation and apoptotic cell death.
CB1 receptors are expressed in the central nervous system as well as the pituitary gland, immune cells, reproductive tissues, gastrointestinal tissues, heart, lungs, urinary bladder, and adrenals (reviewed by Berdyshev1). In contrast, CB2 receptors are found primarily in immune cells, including T cells, B cells, natural killer cells, macrophages, neutrophils, and mast cells.33 34 Thus, the selective expression of CB2 receptors on the immune cells provides a unique opportunity to target malignancies of the immune system by using CB2 agonists to induce apoptosis and thereby provide new avenues to treat such cancers. The advantage in using CB2-selective agonists also stems from the fact that such a treatment is devoid of the psychotropic effects that are characteristic of CB1 agonists. Thus, it is possible to synthesize new ligands for CB2 receptors and test them for their efficacy against tumors of immune origin. It should be noted that in the current study, we randomly selected a few murine and human tumor-cell lines of immune origin that were all found to be sensitive to cannabinoid-induced apoptosis. Recently, however, we identified a human cell line that was resistant, and further studies are in progress to address whether this cell line lacks physical or functional cannabinoid receptors and/or signaling molecules that trigger apoptosis.
The dose of THC that induced apoptosis in vitro in the current study was found to be 10 μM or greater using serum-containing medium and 3 μM or higher in serum-free medium. Similar observations were made by others who also noted that THC was less effective in inducing cell death in the presence of serum.17This is believed to be the result of direct interactions between serum proteins, such as albumin, and cannabinoids.16 The doses of THC used in vitro in the current study were pharmacologically relevant because in an earlier study, rats injected with 50 mg/kg THC were shown to exhibit 10 μM THC in the serum within 10 hours of administration.35 Also, in these studies, mice were given as high as 500 mg/kg 5 times a week for 2 years. Interestingly, despite such high doses, the survival of dosed rats was higher than in controls. Also, the incidence of a wide range of cancers in mice and rats treated with THC was reduced in a dose-dependent manner.35 In the current study, we also observed that cannabinoid agonist WIN55212 failed to induce apoptosis, similar to the findings of Ruiz et al10 in the human prostate-tumor model. The reason why WIN55212 fails to induce apoptosis is not clear. However, we speculate that it is due to differences in the structure and binding abilities to the cannabinoid receptors when compared with the classic cannabinoids such as THC and HU-210.20 In most previous studies, the effect of THC or other cannabinoids in inducing apoptosis in nonlymphoid tumor-cell lines was seen only after exposure for 2 or more days.9 10 12 13 In contrast, in the current study, we were able to demonstrate marked induction of apoptosis in lymphoid tumors as early as 4 hours following culture with THC. These data suggest that lymphoid tumors may be highly sensitive to THC-induced apoptosis. Anandamide was shown recently to induce apoptosis in human neuroblastoma (CHP100) and lymphoma (U937) cells.18 These authors demonstrated that anandamide-induced apoptosis was independent of cannabinoid receptors and was induced through vanilloid receptors. In the current study, we also observed that anandamide was effective at inducing apoptosis in lymphoid cell lines that were screened. Further studies are in progress to determine the involvement of vanilloid or cannabinoid receptors in anandamide-induced apoptosis.
In the current study, we observed that THC was able to induce apoptosis in tumor cells not only in vitro, but also in vivo. Furthermore, THC was effective in reducing the tumor load, prolonging the mean survival time of tumor-bearing mice, as well as curing a significant proportion of such mice. Because THC is immunosuppressive and EL-4 is an immunogenic tumor,36 it is possible that the immunosuppressive effects of THC may have interfered with the host’s antitumor immunity, which may account for a lower percentage of cures. Thus, further manipulations of the dose of THC that would induce significant apoptosis without causing significant suppression of antitumor immunity may lead to development of a treatment regimen that may cure a larger percentage of tumor-bearing mice. Such studies are currently in progress.
The current study demonstrates that targeting CB2 receptors to induce apoptosis may constitute a novel approach to treating malignancies of the immune system. The advantage in using CB2 receptor agonists is that they do not exhibit psychoactive properties. Also, because CB2 receptors are expressed exclusively on immune cells, use of CB2 receptor agonists will not be toxic to nonimmune cells. Thus, further research on CB2 receptor agonists to target transformed immune cells could lead to discovery of a new class of highly selective anticancer agents.

One individual can begin a movement that turns the tide of history. Martin Luther King in the civil rights movement, Mohandas Ganhi in India, Nelson Mandela in South Africa are examples of people standingup with courage and non-violence to bring about needed changes. ~Jack Canfield
Cannabinoid receptor-mediated apoptosis induced by R(+)-methanandamide andWin55,212-2 is associated with ceramide accumulation and p38 activation in mantle cell lymphoma.
…Discussion
Various kinds of cannabinoids have been shown to induce apoptosis in human leukemia and lymphoma cell lines via CB2, the cannabinoid receptor normally expressed in the immune system (McKallip et al., 2002a). We have demonstrated recently that cannabinoid receptor ligands induce growth reduction and apoptosis in MCL (Flygare et al., 2005), a B-cell lymphoma that expresses high levels of both CB1 and CB2 (Islam et al., 2003). In the current study, we examined the role of each receptor, and investigated involvement of MAPK signaling and ceramide accumulation in the induction of apoptosis by two synthetic cannabinoids. Together, our data suggest that targeting CB1 and CB2 receptors may constitute a novel approach to treating MCL.
First, we showed that both R(+)-MA and Win55 induced apoptosis in MCL. The effect was more pronounced after treatment with the CB1 selective agonist R(+)-MA compared with the mixed CB1/CB2 agonist Win55. Programmed cell death in response to Win55 treatment has previously been observed in skin cancer (Casanova et al., 2003) and prostate cancer cells (Sarfaraz et al., 2005) that express both CB1 and CB2. R(+)-MA has recently been shown to induce apoptosis in a cannabinoid receptor independent manner in glioma cells (Hinz et al., 2004). In MCL, caspase-3 activation induced by either R(+)-MA or Win55 was completely reversed by preincubation with 10 nM SR141716A or SR144528. At this dose, the inhibitors act merely as neutral antagonists to CB1 and CB2, respectively, whereas roles as inverse agonists have been suggested at higher doses (Pertwee, 2005). Therefore, our current data suggest that R(+)-MA and Win55, irrespective of differences in CB1/CB2 selectivity, induce caspase-3 mediated apoptosis in MCL cells via ligation of both cannabinoid receptors. A similar requirement of ligation of both CB1 and CB2 has been observed after treatment of dendritic cells with Δ9-tetrahydrocannabinol (Do et al., 2004).
![]() Endogenous and synthetic cannabinoids exert antiproliferative and proapoptotic effects in various types of cancer and in mantle cell lymphoma (MCL). …Together, our results suggest that therapies using cannabinoid receptor ligands will have efficiency in reducing tumor burden in malignant lymphoma overexpressing CB1 and CB2.
Endogenous and synthetic cannabinoids exert antiproliferative and proapoptotic effects in various types of cancer and in mantle cell lymphoma (MCL). …Together, our results suggest that therapies using cannabinoid receptor ligands will have efficiency in reducing tumor burden in malignant lymphoma overexpressing CB1 and CB2.
Potentiation of cannabinoid-induced cytotoxicity in cancer cell modulation of ceramide metabolism
Abstract
Ceramide levels are elevated in Mantle Cell Lymphoma cells following treatment with cannabinoids. Here, we investigated the pathways of ceramide accumulation in the MCL cell line Rec-1 using the stable endocannabinoid analogue R(+)-methanandamide (R-MA). We further interfered with the conversion of ceramide into sphingolipids that promote cell growth. Treatment with R-MA led to increased levels of ceramide species C16, C18, C24 and C24:1and transcriptional induction of ceramide synthases (CerSs) 3 and 6. The effects were attenuated using SR141716A, which has high affinity to cannabinoid receptor 1 (CB1). The CB1-mediated induction of CerS3 and CerS6 mRNA was confirmed using Win-55,212-2. Simultaneous silencing of CerS3 and CerS6 using siRNA abrogated the R-MA-induced accumulation of C16 and C24. Inhibition of either of the enzymes serine palmiotyl transferase, ceramide synthase, and dihydroceramide desaturase within the de novo ceramide pathway reversed ceramide accumulation and cell death induced by R-MA treatment. In order to enhance the cytotoxic effect R-MA, sphingosine kinase-1 (SK-1) and glucosylceramide synthase (GCS), enzymes that convert ceramide to the pro-proliferative sphingolipids sphingosine-1-phospate and glucosylceramide, respectively, were inhibited. Suppression of either enzyme using inhibitors or siRNA potentiated the decreased viability, induction of cell death and ceramide accumulation induced by R-MA treatment. Our findings suggest that R-MA induces cell death in MCL via CB1-mediated upregulation of the de novoceramide synthesis pathway. Furthermore, inhibition of SK-1 and GCS potentiated ceramide accumulation and cell death induced by R-MA. This is the first study were the cytotoxic effect of a cannabinoid is enhanced by modulation of ceramide metabolism.
INTRODUCTION
Ceramide accumulation is a widely described event in cancers after various treatments [1]. C16-Ceramide is described as one of the major ceramide sub-species whose levels are elevated during apoptosis induced by various agents [2]. For instance, C16 ceramide, generated de novo, was accumulated during androgen ablation in the prostate cell line LNCaP [3]. Both C16 and C24ceramide accumulated during BcR crosslinking in Ramos cells [4, 5]. When ceramide species C18 was specifically induced in UM-SCC-22A cells (sqaumous cell carcinoma of hypopharynx) by overexpression of CerS1, cell growth was inhibited [6].
In previous publications [7–9] we and others observed that induction of ceramide accumulation by cannabinoids leads to apoptosis in MCL, glioma and pancreatic cancer. The signaling leading to cell death was blocked by inhibition of ceramide synthase with Fumonisin B1 (FB1). In MCL, the accumulation of ceramide was mediated through the cannabinoid receptors type 1 (CB1) and type 2 (CB2) which are overexpressed on MCL cells, while control cells lacking the receptors remained unaffected.
There are six species of ceramide synthases (CerSs), and several iso-forms have been described [2]. CerSs 1–6 synthesize ceramides of varying chain length [2]. When CerS3 was overexpressed in HEK-293T cells, an increased production of C18 and C24 ceramide species was observed [10], while overexpression of CerS6 showed that the enzyme preferably synthesized the long chain ceramide species C14 and C16 ceramide [11].
CerSs can act through two different pathways, as they are involved in both de novo synthesis of (dihydro)ceramide as well as regeneration of ceramide from sphingosine in the salvage/recycling pathway, see Fig 1. Several enzymes are involved in the de novo synthesis of ceramide which starts with the precursors L-serine and palmitoyl-CoA. Their conversion into 3-ketosphinganine is catalyzed by Serine Palmitoyl Transferase (SPT) [12]. Further downstream, sphinganine is acylated to dihydroceramide by ceramide synthase (CerS). The dihydroceramide is desaturated by dihydroceramide desaturase (DEGS) to ceramide [13]. On the other hand, in the salvage/recycling pathway, CerSs act on sphingosine that is generated from the breakdown of complex sphingolipids. Since FB1 inhibits CerS, its actions do not distinguish between the activation of the de novopathway vs. the operation of the salvage pathway. Thus, it became important to determine the specific pathway activated by cannabinoids.
..DISCUSSION
Ceramide is known to function as a second messenger in different cellular processes, e.g. induction of apoptosis and differentiation, and its accumulation can be induced by a variety of stimuli [16]. Cannabinoids have been shown to induce ceramide accumulation in pancreatic cancer-, glioma- and leukemia cell lines [8, 9, 34] as well as in MCL [7]. The production of ceramide following treatment with cannabinoids can be caused by either hydrolysis of sphingomyelin or de novo ceramide synthesis [35, 36]. In the current study, we have shown accumulation of ceramide species C16, C18, C24 and C24:1 in the MCL cell line Rec-1 after long-term stimulation with the stable endocannabinoid analogue R-MA. Ceramides exhibit a tissue-dependent bias for amide-linked fatty acids, characterized by chain length, degree of saturation and degree of hydroxylation. C16 ceramide is known to be most abundant in fibroblasts, endothelial cells and immune cells [37, 38]. In accordance with the present study using a cell line of B-cell origin, C16 and C24 ceramide species were accumulated after BcR crosslinking in the B-cell line Ramos [4, 5].
It has been shown previously that cannabinoids can induce apoptosis via de novo synthesis in C6 glioma cells [36]. In the same study, increased activity of SPT was the rate-limiting step in ceramide synthesis de novo [39]. Here, we observed upregulation of messages for ceramide synthases within the same pathway. The six known forms of CerSs are active in two processes, acylating either dihydrosphingosine to form dihydroceramide or sphingosine to form ceramide. After stimulation with R-MA, the Rec-1 cells overexpressed CerS3 and CerS6 two and 2,6 times respectively. CerS6 is a major CerS that is expressed at high levels in a variety of tissues [11], and is associated with synthesis of ceramide species C14 and C16. CerS3 is regarded as a minor CerS that has mainly been described in skin and testis [10, 40], and has been shown to synthesize the longer ceramide species C18, C20 and C24 [10, 11]. The induction of CerS3 after stimulation with cannabinoids is intriguing, since database searches show that CerS3 expression is rarely present in lymphoma tissue, while CerS6 expression is present in a majority of the lymphomas (Omnibus GEO database).
Silencing of either CerS3 or CerS6 using siRNA had no effect, while simultaneous knockdown abrogated the accumulation of C16, C18 and C24. Our results suggest that CerS3 and CerS6 may have redundant functions in the production of ceramide species, and indicate that CerS6 and CerS3 are not restricted to the synthesis of C14 and C16 or longer ceramide species, respectively. Similarly to the effects on ceramide accumulation, simultaneous suppression of CerS3 and CerS6 had a reproducible, but non-significant inhibitory effect on R-MA-induced cell death. Generally, the induction of ceramide accumulation and cell death following R-MA treatment was greater in untransfected compared to transfected cells, as observed when comparing Fig 3 and and4d4d or Fig 8a and 8b, respectively. Thus, electroporation itself is likely to affect the background levels of ceramides and DNA fragmentation, rendering the effects of R-MA less pronounced.
The CB1 antagonist SR1 completely inhibited the R-MA-induced accumulation of ceramide species C16, C18 and C24 and the upregulation of CerS3 and CerS6 following treatment with R-MA or Win55. SR2, an antagonist to CB2, partially inhibited the increase in C16, C18 and CerS3, while the inhibitory effects on the accumulation of other ceramide species and on the upregulation of CerS6 were not significant. Given the selectivity of R-MA towards CB1, it may seem surprising that SR2 counteracted the induction of ceramide accumulation and the upregulation of CerSs. These results are in accordance with our earlier studies showing that antagonists to either CB1 or CB2 attenuated cell death induced by R-MA in MCL and other lymphomas expressing both receptors [7,41]. At the doses used, it is possible that R-MA acts as an agonist also to CB2, despite its much higher affinity to CB1. Alternatively, the binding of SR2 to CB2 induces changes downstream of the receptor that affect the signaling via CB1.
We have previously blocked cell death in Rec-1 cells by inhibiting CerSs with FB1 [7]. The same inhibitor has been used to prevent induction of ceramide accumulation by cannabinoids in glioma cells [42]. However, CerSs can act in two pathways; both in regeneration of ceramide from sphingosine and in de novo synthesis, see Fig 1. To exclude the involvement of SL degradation, we here added inhibitors to enzymes that are active only in the de novo pathway; SPT and DEGS. Dbaibo et al. [43] have shown that inhibition of SPT with myriocin abrogates cell death induced by arsenic trioxide in T-cell leukemia and lymphoma. In the present study, inhibition of either of three different enzymes, SPT, CerS and DEGS, led to an abrogation of ceramide accumulation and suppression of cell death in MCL (Fig 1). This fortifies the role of de novosynthesis in the responses to R-MA.
It has previously been suggested that ceramide is the active mediator of apoptosis, while dihydroceramide is merely an inactive precursor to ceramide [14]. In these studies, exogenous ceramide and dihydroceramide have been added to cells or mitochondria [44] [45]. In contrast, high levels of dihydroceramide were observed in HL-60 leukemia cells prior to cell death, [46] and SMS-KCNR cells were cell cycle arrested when DEGS was inhibited [47]. In our MCL cells, the inhibition of DEGS using C8-CPPC caused accumulation of dihydroceramide species (data not shown) and inhibited the induction of cell death by R-MA. This supports the theory that dihydroceramide species cannot induce apoptosis when the transformation to ceramide is inhibited.
The above conclusion on the central role of ceramide suggested that the cell death-promoting effects of R-MA could be enhanced by inhibiting the transformation of ceramide into species with opposing effects. Inhibition of both SK-1 and GCS potentiated the effects of R-MA on viability and cell death. French et al [33] have shown that SKI II is a specific inhibitor to S-1-P formation, which also inhibits tumor growth in vivo. In view of the significant potentiation of the R-MA-induced effects observed using SKI II in vitro, future in vivo studies using a combination of R-MA and SKI II in mice xenotransplanted with MCL cells are warranted. To be assured that the effects observed in our experiments were enzyme-specific, GCS and SK1 were silenced using siRNA. In addition to potentiation of the effects induced by R-MA, silencing of SK-1 led to a decrease in cell viability by itself. Taha et al [18] and Sarkar et al [48] have observed that down-regulation of SK-1 by siRNA in MCF-7 cells resulted in a reduction of cell viability. Interference with GCS RNA has also been shown to affect growth of neuroepitelioma [44] and sensitize breast carcinoma cells to cytotoxic drugs [43]. However, the viability of an astrocytoma cell line following treatment with the cannabinoid Δ-9 tetrahydrocannabinol (THC) was not affected by inhibitors or siRNA against GCS [49]. In our cells, knockdown of SK-1 led to significant potentiation of R-MA-induced C16 accumulation, whereas the accumulation of both C16 and C18 was potentiated using siRNA against GCS. Instead, the levels of C24 and C24:1 remained unaltered. It is possible that these ceramide species are metabolized by other ceramide-converting enzymes, e.g. sphingomyelin synthase or ceramide kinase [1].
In conclusion, we have shown that induction of CerSs regulate de novo ceramide synthesis in response to the stable endocannabinoid analogue R-MA in MCL cells. The effect of inhibition of DEGS supports earlier studies showing that ceramide, and not dihydroceramide, is the active mediator of apoptosis. Moreover, inhibition of enzymes that convert ceramide to growth-promoting sphingolipid species potentiated the ceramide accumulation and cell death induced by R-MA in MCL. Cannabinoids have been suggested as a new non-toxic therapeutic option for cancer treatment [50]. This is the first study showing that the cytotoxic effect of a cannabinoid can be enhanced by modulation of ceramide metabolism. The results suggest that interference with ceramide conversion may provide a tool to enhance the targeted cell death-promoting effects of cannabinoids in MCL and other malignant lymphomas overexpressing the CB1 receptor.
![]() Recent studies have documented the potential antitumorigenic properties of CBD in the treatment of various neoplasms, including breast cancer,24 lung cancer,25 bladder cancer,26 glioblastoma,27 and leukemia.28 CBD induces these effects through a variety of mechanisms and signaling pathways.
Recent studies have documented the potential antitumorigenic properties of CBD in the treatment of various neoplasms, including breast cancer,24 lung cancer,25 bladder cancer,26 glioblastoma,27 and leukemia.28 CBD induces these effects through a variety of mechanisms and signaling pathways. 
..Although CBD is reportedly effective against various tumors, its molecular mechanism of action is not fully characterized. CBD is cytotoxic to gliomas and inhibits tumor cell migration in vitro (7–9). In addition, CBD induces apoptosis in human leukemia cell lines by activating classical caspase pathways, and enhancing NOX4 and p22 (PHOX) function (10). A recent study reports that CBD inhibits breast cancer growth (11) and downregulates ID1, a regulator of metastasis in breast cancer cell lines (12). Furthermore, CBD, in conjunction with THC, induces programmed cell death (PCD) in glioma cells (13).
“These data enhance the desirability of CBD as an anticancer agent, while minimizing damage to normal tissue.”
Apoptosis can be induced either by the intrinsic pathway (which is mediated by the mitochondria) or signaling from death receptors at the cell surface (the extrinsic pathway; refs. 14, 27). We found that CBD induced the activation of caspase-8, the generation and translocation of t-BID to the mitochondria, the release of cytochrome c and SMAC into the cytosol, and increased levels of Fas-L. These data support our hypothesis that CBD induces PCD (programmed cell death).
Recent publications show that ROS play a central role in the CBD-induced death of glioma and Jurkat cells (8, 10). Consistent with these studies, we observed CBD-enhanced ROS generation in breast cancer cells. Moreover, when we limited ROS levels with TOC, CBD-induced expression of protein markers for both apoptosis and autophagy decreased, and levels of apoptosis decreased, suggesting that ROS play a critical role in the CBD-induced PCD of breast cancer cells.
We explored the relationship between CBD-induced apoptosis and autophagic cell death by blocking each form of PCD with specific caspase and autophagy inhibitors. Caspase inhibition decreased levels of CBD-induced apoptosis and protein markers in breast cancer cells, whereas inhibiting autophagy enhanced levels of CBD-induced apoptosis and protein marker expression. These data suggest that blocking CBD-induced autophagy may affect a compensatory increase in apoptosis as an alternative means of PCD. We hypothesize that CBD may mediate this ratio between apoptosis and autophagy in CBD-induced PCD via beclin-1. Beclin-1 can block autophagy when it forms a protein complex with Bcl-2 (34). However, if beclin-1 is cleaved, this complex dissociates and autophagy is induced (37). Moreover, this cleavage product translocates to the mitochondria and induces apoptosis by enhancing the release of cytochrome c (36). We observed a CBD-induced translocation of beclin-1 to the mitochondria which suggests that a potential mechanism by which CBD affects the balance between autophagy and apoptosis in breast cancer cells is by inducing the cleavage and translocation of beclin-1.
In summary, we showed that CBD, a plant-derived cannabinoid, preferentially kills breast cancer cells by inducing ER stress, inhibiting mTOR signaling, enhancing ROS generation, and mediating a complex balance between autophagy and mitochondria-mediated apoptosis in MDA-MB-231 breast cancer cells. These findings support the continued exploration of CBD as an alternative agent for breast cancer treatment.
Neil Young – Helpless (Live At Massey Hall – 1971)


http://nslegislature.ca/legc/statutes/childfam.htm http://worldtruth.tv/75-of-physicians-in-the-world-refuse-chemotherapy-for-themselves/ http://www.cheofoundation.com https://www.freedomwares.ca/cannabinoids-therapeutic-agents-cancer-current-status-future-implications/ http://www.cancer.ca/en/cancer-information/diagnosis-and-treatment/chemotherapy-and-other-drug-therapies/chemotherapy/side-effects-of-chemotherapy/?region=on#ixzz3EOQaPSyj
http://www.georgiatoons.com

