

Jump to section
Visual Overview

Abstract

Molecular Pharmacology Vol. 90, Issue 5 1 Nov 2016
The cannabinoid 1 receptor (CB1R) is one of the most abundant G protein–coupled receptors (GPCRs) in the central nervous system, with key roles during neurotransmitter release and synaptic plasticity. Upon ligand activation, CB1Rs may signal in three different spatiotemporal waves. The first wave, which is transient (<10 minutes) and initiated by heterotrimeric G proteins, is followed by a second wave (>5 minutes) that is mediated by β-arrestins. The third and final wave occurs at intracellular compartments and could be elicited by G proteins or β-arrestins. This complexity presents multiple challenges, including the correct classification of receptor ligands, the identification of the signaling pathways regulated by each wave, and the underlying molecular mechanisms and physiologic impacts of these waves. Simultaneously, it provides new opportunities to harness the therapeutic potential of the cannabinoid system and other GPCRs. Over the last several years, we have significantly expanded our understanding of the mechanisms and pathways downstream from the CB1R. The identification of receptor mutations that can bias signaling to specific pathways and the use of siRNA technology have been key tools to identifying which signaling cascades are controlled by G proteins or β-arrestins. Here, we review our current knowledge on CB1R signaling, with particular emphasis on the mechanisms and cascades mediated by β-arrestins downstream from the CB1R.
Introduction
Functional selectivity, also called ligand or receptor bias, is the ability of ligands to activate a subset of the full repertoire of signaling cascades available to individual G protein–coupled receptors (GPCRs) (Urban et al., 2007). This concept, based on numerous analytical observations from different laboratories, challenged our classic definition of a ligand’s intrinsic efficacy (i.e., the property of a ligand/receptor pair to elicit the full set of biologic responses). Functional selectivity also provided a novel conceptual framework and therapeutic opportunities to pharmacologically control GPCR function (Ariens, 1954; Kenakin, 2004), which resulted in the realization that signaling from individual GPCRs is even more complex than initially proposed and that receptor signaling is in fact pluridimensional (Galandrin et al., 2007; Kenakin, 2011; Luttrell, 2014). Elucidation of GPCR pluridimensionality has led to the rational search for more effective therapeutic agents and the systematic exploration of the signaling pathways and molecular mechanisms underlying functional selectivity. With the ability to specifically abrogate desired proteins, either genetically or chemically, while analyzing downstream effectors, our current knowledge of these events has increased exponentially; however, we are still far from having a complete picture of the events that follow GPCR activation that lead to biologic responses. Assisted by the development of powerful computational tools for structure/function analysis, additional relevant pieces of this intricate jigsaw puzzle are realized almost daily with the continuous description of GPCR crystal structures and their associated proteins (Katritch et al., 2013; Maudsley et al., 2013; Johnston and Filizola, 2014; Shukla et al., 2014a).
In this review, we discuss functional selectivity of the cannabinoid 1 receptor (CB1R), one of the most highly expressed GPCRs in the central nervous system. First, we review the different signaling waves characterizing functional selectivity of the CB1R that are mediated by G proteins and β-arrestins. Then we focus the discussion on ligand bias toward β-arrestins and the underlying molecular mechanisms. We discuss the in vivo data from wild-type, knockout, and knock-in mice and conclude by highlighting intriguing problems and suggesting areas where further research is needed to understand the physiologic roles and therapeutic potential of β-arrestin–mediated signaling of the CB1R.
The CB1R is one of the most abundant GPCRs in the central nervous system, with expression levels ranging between 0.5 and 7 pmol/mg protein in numerous areas of the rat brain (Herkenham et al., 1991; Mackie, 2008; Marsicano and Kuner, 2008). CB1R localization in neuronal cells is highly polarized to axons and presynaptic sites, where they control synaptic neurotransmitter release and neuronal function (Howlett et al., 1990; Mackie, 2008; Castillo et al., 2012). They are activated by their endogenous ligands (endocannabinoids), such as anandamide and 2-arachidonyl glycerol (2-AG), and they are also activated by tetrahydrocannabinol (Δ9-THC), the main psychoactive ingredient in marijuana, that has been linked to multiple physiopathological conditions. The pharmacologic regulation of the CB1R has been proposed as a therapeutic strategy for many neuropsychiatric disorders ranging from anxiety and stress to neurodegenerative disease and epilepsy (Howlett et al., 2002; Howlett, 2005; Mackie, 2006; Lutz et al., 2015).
CB1 Receptor Signals in Three Different Waves
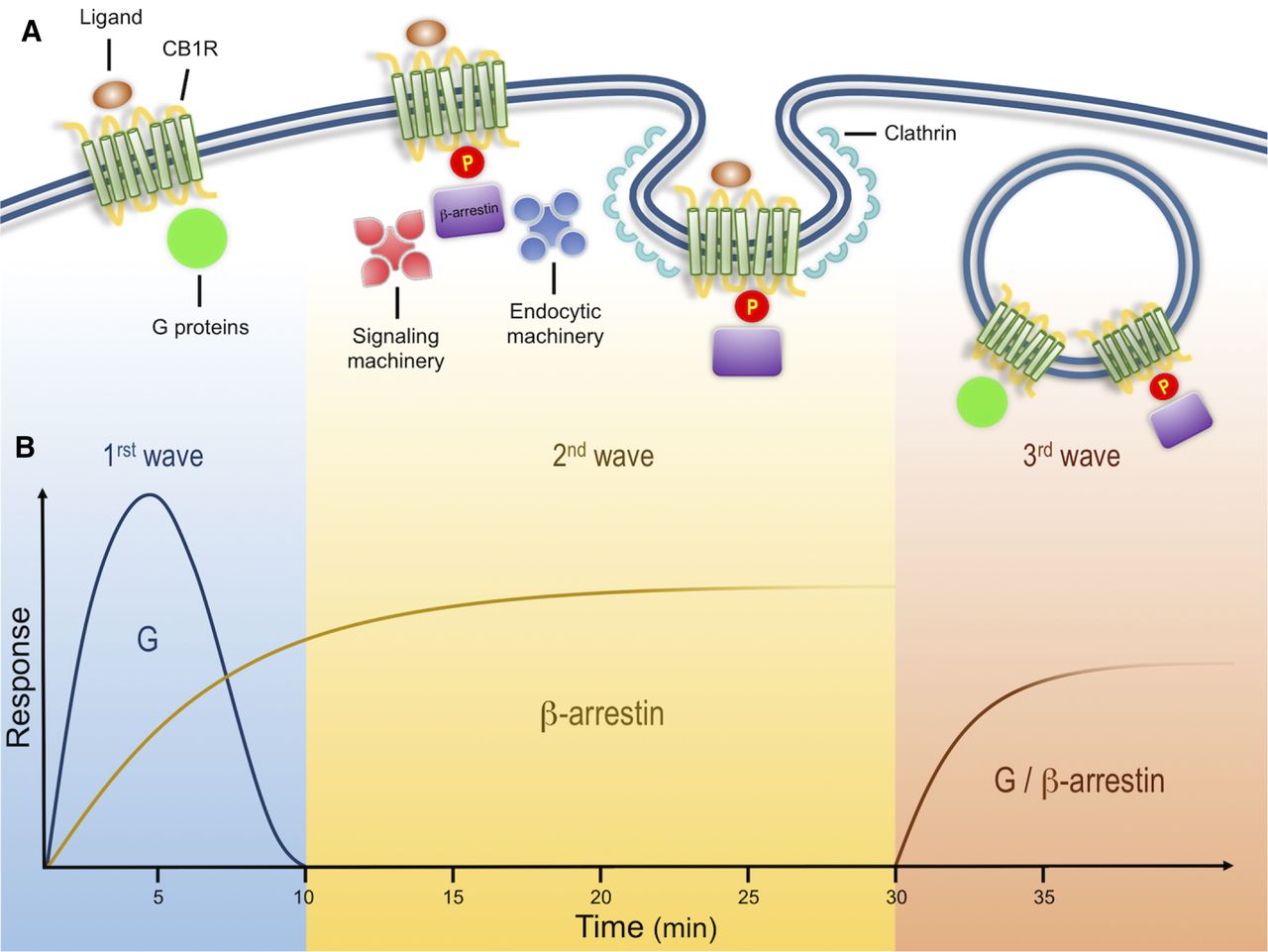
Biochemical analysis indicates that CB1Rs, like multiple other GPCRs, such as angiotensin type 1 receptor and type 1 parathyroid hormone-related protein receptor, can signal in three distinct spatiotemporal waves (Luttrell and Gesty-Palmer, 2010; Lohse and Calebiro, 2013). An initial wave mediated by heterotrimeric Gαi/o proteins begins after ligands bind receptors at the plasma membrane, leading to a rapid decrease in cAMP levels, a decrease in Ca2+conductance, and an increase in K+ conductance (Fig. 1) (Howlett et al., 2004). CB1Rs present significant functional selectivity at the G protein level. CB1Rs couple mainly to heterotrimeric Gi/o but also to other G proteins (Glass and Northup, 1999; Varga et al., 2008; Bosier et al., 2010). This promiscuity has been extensively characterized utilizing in vitro assays in different cell lines, further emphasizing the generalized notion that the cellular environment is highly relevant during CB1R signaling; careful consideration must be taken when interpreting results obtained from heterologous systems where different protein levels of receptor/signaling molecules can have a profound effect on their function (Bosier et al., 2010; Atwood et al., 2011; Straiker et al., 2012).
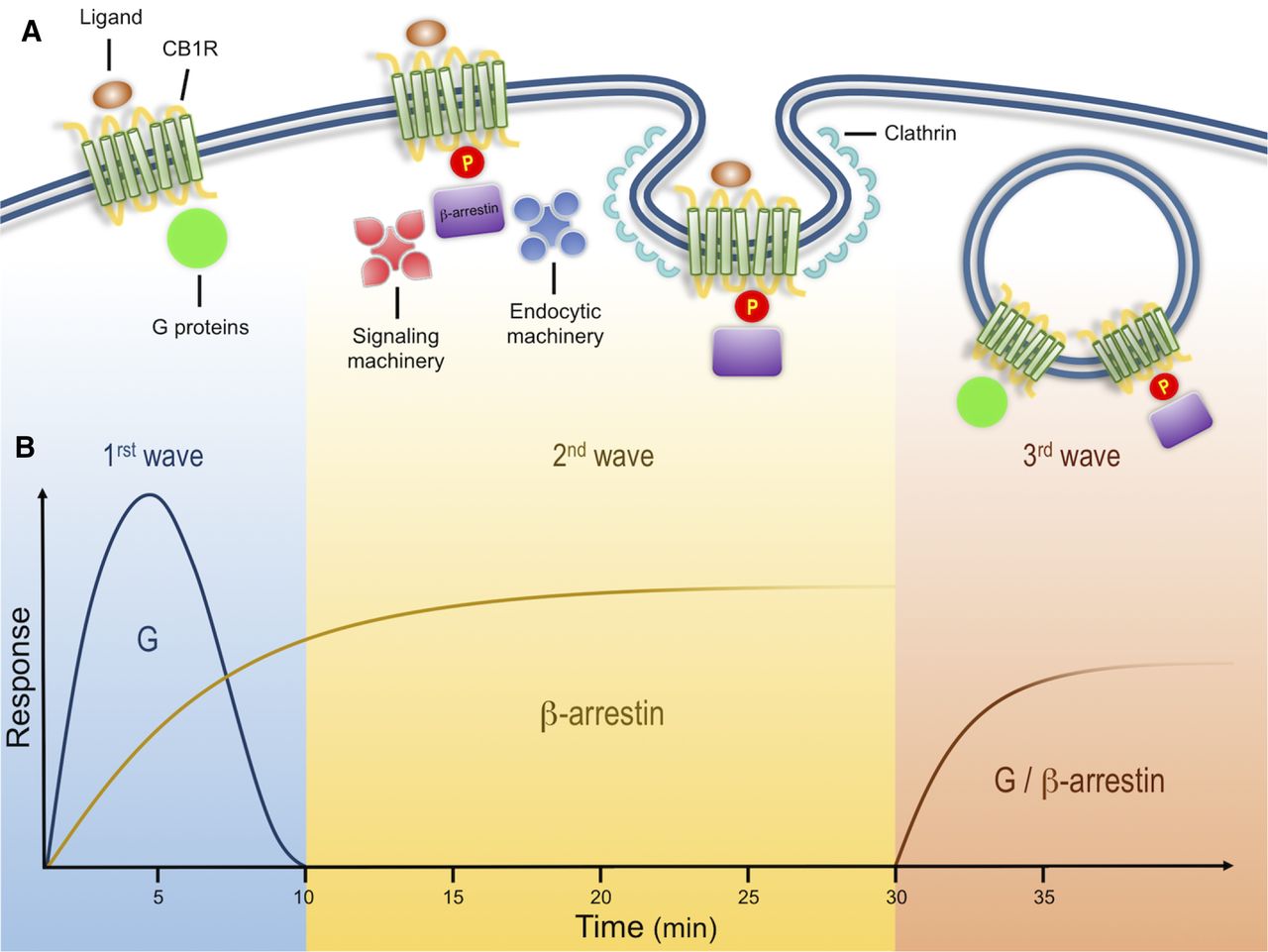
Cannabinoid receptor signals in three waves. (A) Activation of CB1Rs results in the modulation of multiple cellular responses through three distinct signaling waves. The first wave, mediated by G proteins, is observed within seconds and up to few minutes after receptor activation. Receptor activation also results in phosphorylation by GRKs. This post-translational modification leads to receptor desensitization and the recruitment of β-arrestins, scaffold proteins of the endocytic machinery that initiate clathrin-mediated endocytosis. In addition to the endocytic machinery, receptor bound β-arrestins can also recruit and activate signaling proteins, resulting in a second signaling wave with distinct kinetic and signaling profile. These events are initiated at the plasma membrane and can continue after receptor endocytosis into intracellular compartments. After receptor internalization, a third signaling wave has been described that is characterized by the activation of effectors associated with both G proteins and β-arrestins. (B) Proposed time course of G protein– and β-arrestin–mediated responses. G protein signaling has a fast initial response, whereas β-arrestins are somewhat slower but sustained over time. Kinetics of third waves can be initiated within minutes (modified from Luttrell and Gesty-Palmer, 2010).
Ligand-induced receptor phosphorylation results in receptor desensitization and the recruitment of the scaffold β-arrestins (Jin et al., 1999; Kouznetsova et al., 2002). β-arrestins, while hindering G-protein signaling, act as scaffold proteins for the endocytic machinery and signaling molecules such as the mitogen-activated protein family of kinases and initiate the second wave of signaling at the cell surface (Ahn et al., 2013; Flores-Otero et al., 2014). This review focuses on this wave and the mechanisms underlying these events below.
A final third wave emerges from receptors localized at intracellular compartments, such as endosomes and lysosomes. Native CB1Rs are particularly enriched at intracellular compartments in Neuro2A cells and primary hippocampal cultures when analyzed by immunostaining and discontinuous sucrose gradients (Rozenfeld and Devi, 2008). By combining the lipophilic agonist WIN-55212-2 [(11R)-2-methyl-11-(morpholin-4-ylmethyl)-3-[(naphthalen-1-yl)carbonyl]-9-oxa-1-azatricyclo[6.3.1.0^{4,12}]dodeca-2,4,6,8(12)-tetraene] with the receptor blocker peptide hemopressin, which does not cross the plasma membrane, signaling from intracellular native receptors was shown to stimulate extracellular signal-regulated protein kinase (ERK) phosphorylation in Neuro2A cells. Indirect support of intracellular signaling was further presented by coimmunoprecipitation of G proteins and CB1Rs from endosomal compartments isolated from these cells (Rozenfeld and Devi, 2008). More recently, calcium release from intracellular stores was demonstrated by injecting ananadamide into human embryonic kidney cells transfected with CB1Rs, further suggesting a signaling wave from receptors localized in intracellular compartments (Brailoiu et al., 2011). Careful consideration should be taken, however, when analyzing receptor signaling and trafficking, particularly when comparing heterologous systems, different cellular models, and ligands at saturating or high concentrations. When trying to understand receptor function, factors that should be taken into consideration include differences in receptor and accessory protein expression levels, their cellular localization, ligand bias, and ligand on/off rates among others.
Are these signaling waves relevant in vivo? What are their possible biologic roles? Are they present in some, but not all, cells? We are at the beginning of a new era of GPCR pharmacology, and these questions must be addressed to help the rational design of new and improved therapeutics.
Multifaceted β-Arrestins
Four highly homologous β-arrestin isoforms have been described in mammals. Arrestin 1 and arrestin 4 (visual arrestins) are expressed only in the retina, and arrestin 2 and 3 (referred in this review as β-arrestin 1 and β-arrestin 2, respectively) are expressed ubiquitously (Gainetdinov et al., 2004; Gurevich and Gurevich, 2006; Premont and Gainetdinov, 2007). β-arrestins were initially identified in the late 1980s and early 1990s as key proteins during the inactivation or “arrest” of ligand activated GPCRs (Pfister et al., 1985; Benovic et al., 1987; Lohse et al., 1990; Schmid and Bohn, 2009), and a second critical role for β-arrestins during the ligand-induced receptor internalization was discovered soon after. The β-arrestin C terminus binds directly to clathrin and the adaptor protein 2, thus working as a scaffold for the endocytic machinery leading to the removal of desensitized receptors from the cell surface via clathrin-mediated endocytosis (Goodman et al., 1996; Laporte et al., 1999). More recently, a third function was described; β-arrestin recruitment to phosphorylated receptors initiates a G protein–independent wave of signaling that results in the activation of multiple effectors including ERK, c-Jun N-terminal kinase (JNK), and SRC proto-oncogene nonreceptor tyrosine kinase (Src), among others (Gurevich and Gurevich, 2006; DeWire et al., 2007; Luttrell and Gesty-Palmer, 2010; Shenoy and Lefkowitz, 2011). Not surprisingly, β-arrestin function as signaling facilitator is specific and dependent on the type of receptor, ligand, and cellular environment (Whalen et al., 2011; Srivastava et al., 2015).
With the available crystal structures and computational modeling, we can outline the events characterizing the interaction between β-arrestins and activated receptors. It has been postulated that this interaction consists of two differential and sequential steps, initially between the receptor carboxy terminus and the N domain of β-arrestins and later between the receptor transmembrane core and different surface areas at the concave region of β-arrestins (Gurevich and Gurevich, 2004; Shukla et al., 2014b; Kang et al., 2015). Receptor binding results in increased dynamics at the N and C domains of β-arrestins, and this likely affects their interaction with the endocytic and signaling machinery.
Among the possible signaling cascades available to GPCRs, those controlled by β-arrestins have been suggested as good candidates to mediate some of the beneficial effects attributed to current therapeutic compounds. For example, the Food and Drug Administration–approved β-blocker carvedilol (Warne et al., 2008; Tzingounis et al., 2010) and the agonist isoetharine (Drake et al., 2008; Liu et al., 2012) have different patterns of signaling among G protein and β-arrestin–mediated pathways. Interestingly, the antipsychotic drug aripiprazole is highly efficacious at activating signaling cascades mediated by β-arrestins from the dopamine 2 receptors, and this has led to a search for new dopaminergic antipsychotics with improved efficacy (Mailman, 2007; Allen et al., 2011; Urs et al., 2014; Brust et al., 2015). Ligand bias toward β-arrestins has been implicated in several pathologic events, including cardiovascular disorders, pain responses, and some of the behavioral effects associated with cannabis use, which suggests that β-arrestin–mediated signaling components could be potential therapeutic targets (Breivogel et al., 2008; Whalen et al., 2011).
CB1Rs and β-Arrestin–Mediated Signaling
In vivo, the role of β-arrestins as signaling molecules is somewhat elusive. Canonical functions of arrestins, including receptor desensitization, regulation of receptor sensitivity to acute agonists, and regulation of receptor internalization, have been reported (Breivogel et al., 2008; Whalen et al., 2011; Nguyen et al., 2012). β-arrestin 2 knockout mice displayed enhanced antinociceptive responses to acute Δ9-THC and decreased tolerance, similar to the enhanced antinociceptive action observed with opioids in the knockout animals, and more likely associated with reduced receptor desensitization and/or enhanced G protein signaling rather than β-arrestin mediated signaling (Bohn et al., 2000; Nguyen et al., 2012). Somewhat similar results were observed with a knock-in mice where the putative CB1R G protein–coupled receptor kinases (GRK) sites S426/430 were mutated to alanines (Morgan et al., 2014). These mice were more sensitive to acute Δ9-THC, present delayed tolerance, and have reduced receptor desensitization (Morgan et al., 2014). Interestingly, work on β-arrestin 1 knockout mice demonstrated a reduced ability of the full agonist CP 55,940, but not Δ9-THC, to induce antinociception and hypothermia, suggesting either a signaling role of β-arrestin 1 or compensatory actions of β-arrestin 2 on CB1 receptors. These data provide new support to the divergent roles of β-arrestin 1–2 in vivo and suggest that β-arrestin 1 regulates receptor sensitivity in an agonist-dependent manner with no significant effect on the regulation of cannabinoid tolerance (Breivogel and Vaghela, 2015). Substantial effort should be devoted to further distinguish desensitization versus signaling roles of β-arrestins in vivo, particularly for the development of biased therapeutics and for efforts directed toward unraveling β-arrestin function in vivo.
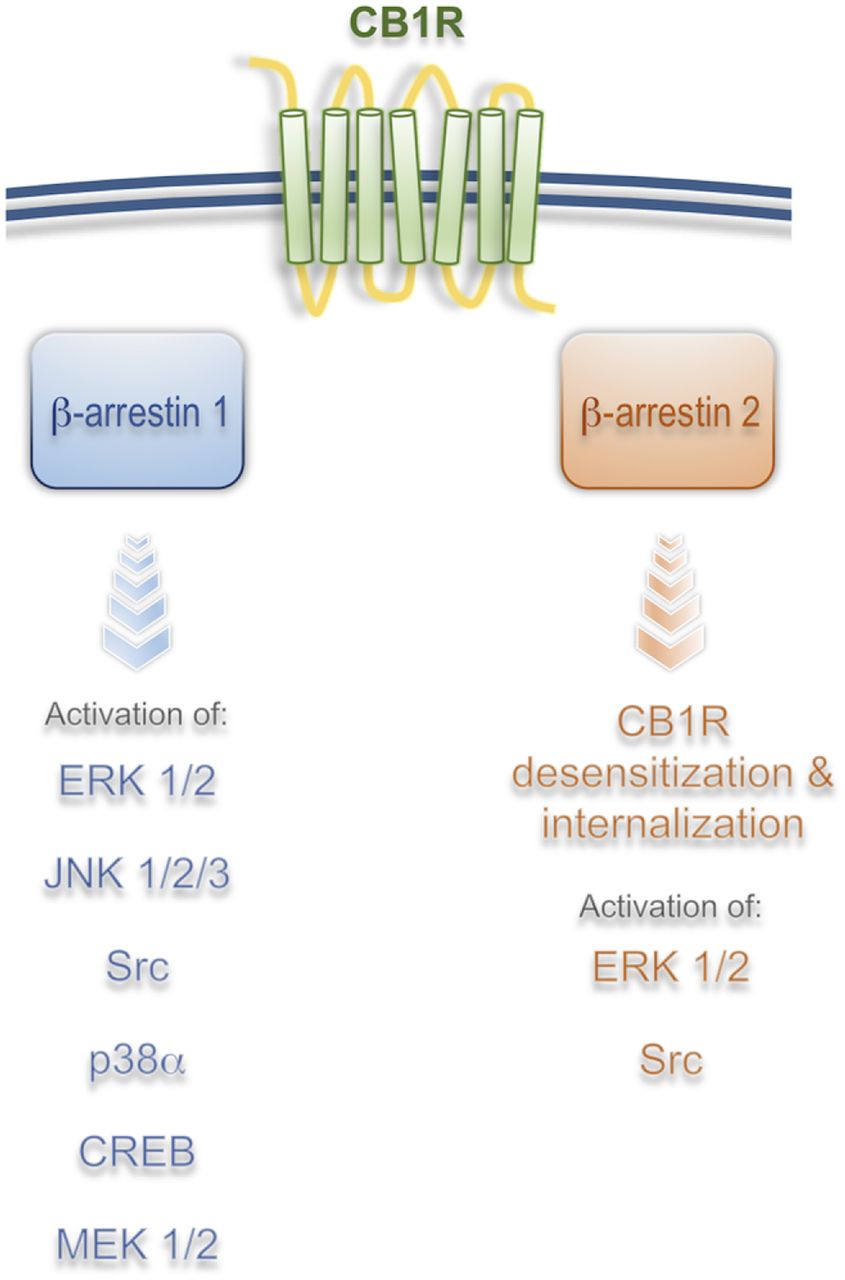
At the molecular level, it was initially suggested that G protein–independent signaling from the CB1R was a possible mechanism for the activation of certain kinases and for the regulation of gene transcription (Jin et al., 1999; Bosier et al., 2008; Daigle et al., 2008; Ahn et al., 2012). Activation of pertussis toxin-insensitive signaling cascades from the CB1R by the ago-allosteric modulator Org27569 (5-chloro-3-ethyl-N-[2-(4-piperidin-1-ylphenyl)ethyl]-1H-indole-2-carboxamide) was demonstrated in hippocampal neurons endogenously expressing CB1Rs (Ahn et al., 2012; Baillie et al., 2013). β-arrestin–mediated signaling downstream from the CB1R was first unequivocally demonstrated in human embryonic kidney cell line HEK293 using a combination of β-arrestin 1/2 siRNA and pertussis toxin treatment (Ahn et al., 2013). In these experiments, ORG27569 elicited strong ERK1/2, Src, and MEK1/2 phosphorylation that were reduced only by β-arrestin 1 siRNA (Ahn et al., 2013). Interestingly, β-arrestin 2 was not involved in the signaling process but in the internalization of the receptor, suggesting distinct roles for these molecules. The combination of pertussis toxin, dominant negative Gαi/o minigenes, and Gβγ-scavenging peptides was later used by Mahavadi et al. (2014) to show that activation of CB1 receptor by anandamide (AEA) in cultured smooth muscle cells results in a GRK5/β-arrestin 1/2 time-dependent activation of ERK1/2 and Src. Interestingly, both β-arrestin 1 and 2 siRNA were effective at reducing ERK1/2 activity (Mahavadi et al., 2014). Differences in the roles of β-arrestin 1/2 among studies are likely a result of different cell systems or different strategies used; however, overlapping, as well as divergent roles of β-arrestins, have been well documented in many cell models, receptors, and tissues (Srivastava et al., 2015) (Fig. 2).
{kind=link}
{kind=link}
{kind=link}
β-Arrestin effectors downstream from CB1R. Brief summary of published results utilizing different cellular and tissue models suggests that β-arrestin 1 mediates most of the signaling whereas β-arrestin 2 mediates receptor desensitization and internalization in vitro and in vivo. Signaling from CB1R/β-arrestin 1 results in regulation of gene transcription and protein synthesis, which suggests that long-term effects of CB1R activation are mediated by β-arrestins (see text for more detail).
Additional characterization of the effect of cannabinoids on the activation of kinases and the regulation of gene expression was investigated using a cell culture model of striatal medium spiny neurons STHdhq7/q7 (Laprairie et al., 2014). In this study, BRET, FRET, and kinase phosphorylation analyses were used to characterize the functional selectivity of six different cannabinoid receptor ligands, including two endocannabinoids and Δ9-THC. 2-AG, Δ9-THC, and CP 55,940 induced a prolonged ERK activation dependent on β-arrestin 1, whereas Akt phosphorylation was mediated by G proteins upon incubation with 2-AG, AEA, and WIN (Laprairie et al., 2014). These results support the notion that functional selectivity of cannabinoid receptor ligands regulates kinase activity selectively. The authors examined whether receptor signaling bias also translates to gene expression using the CB1R as a target gene based on previous findings showing that CB1R mRNA levels are associated with Akt phosphorylation. Results showed that 1µM AEA, 2-AG, or WIN induced an increase in CB1 receptor mRNA levels via Gαi/o proteins in association with the upstream activation of Akt (Laprairie et al., 2014). More recently, the same group reported that CP 55,940 and Δ9-THC preferentially enhanced the recruitment of β-arrestin 1 and reduced cellular viability in a cell model of Huntington disease, supporting the idea that cannabinoids with β-arrestin bias could be detrimental in Huntington disease models (Laprairie et al., 2016).
Recent work showed that 2-AG can induce prolonged (>10 minutes) phosphorylation of ERK1/2, JNK1/2/3, CREB, and P38α via β-arrestin 1 within 5 minutes after ligand incubations (Delgado-Peraza et al., 2016). Interestingly, mutation of putative GRK phosphorylation sites S426/430A resulted in a β-arrestin–mediated signaling-biased receptor. CB1R S426/430A displayed reduced β-arrestin 2 recruitment, associated with a lower internalization rate, and normal β-arrestin 1 recruitment, which is linked to increased β-arrestin 1–mediated signaling (Delgado-Peraza et al., 2016). This result supports the hypothesis that ligands induce specific receptor phosphorylation profiles that result in unique signaling cascades (Fig. 2).
Mechanism Controlling β-Arrestin–Mediated Signaling
Upon ligand binding, GPCRs undergo conformational changes leading to the activation of heterotrimeric G proteins and their effector cascades. These changes in receptor conformation are detected by G protein–coupled receptor kinases (GRKs), resulting in specific phosphorylation patterns at receptor intracellular domains. Quantitative mass spectrometry approaches, combined with phosphospecific antibodies, have shown that ligand-induced receptor phosphorylation is tissue and ligand specific and can be associated with specific signaling cascades; this supports a phosphorylation barcode hypothesis (Butcher et al., 2011; Liggett, 2011; Nobles et al., 2011; Prihandoko et al., 2016). Receptor phosphorylation is recognized by β-arrestins, which are recruited to the plasma membrane and sterically hinder G protein association while initiating β-arrestin–mediated internalization and signaling (Nobles et al., 2011; Liggett, 2011). Data on the CB1R indicate that specific GRKs and phosphorylation sites at the receptor are necessary for β-arrestin–mediated signaling, further supporting the barcode model; however, how phosphorylated receptors transduce their activation into β-arrestin–mediated signaling was not defined until recently (Flores-Otero et al., 2014).
By directly visualizing individual CB1R endocytic events, the ligand modulation of endocytic dwell time, or the time receptors and β-arrestin cluster at the cell surface inside coated pits before their endocytosis was proposed to be a process that controls β-arrestin–mediated signaling (Flores-Otero et al., 2014). Synthetic ligands, such as CP 55,940 or WIN, elicit short dwell times (<120 seconds) and little detectable β-arrestin 1–mediated signaling, whereas 2-AG elicits prolonged dwell times (>120 seconds) and significant β-arrestin 1–mediated signaling (Flores-Otero et al., 2014). Supporting the correlation between dwell times and β-arrestin–mediated signaling, recent data show that β-arrestin–mediated signaling can be increased by inhibiting the internalization of receptors clustered into coated pits while prolonging their interaction with β-arrestins at the cell surface. Interestingly, CB1R endocytic dwell times are strictly dependent on the ligand and can be divided into either short (<120 seconds) or long (>120 seconds) dwell times. This differential response could be used to probe for ligands that promote β-arrestin–mediated signaling.
At the mechanistic level, this work showed that receptor prolonged dwell times are dependent on serines 426/430 (rat sequence conserved in human). Mutation of these sites resulted only in prolonged dwell times that triggered enhanced β-arrestin 1–mediated signaling, reduced β-arrestin 2 recruitment, and decreased receptor internalization rates (Delgado-Peraza et al., 2016). As shown by immunoprecipitation, this interaction between the mutant receptor and β-arrestin 1 is enhanced and continues after internalization into intracellular compartments, a result that led to the use of the S426/430A mutant receptor as a tool to investigate β-arrestin–mediated signaling from the CB1receptor. Data obtained from this receptor indicated that ERK1/2, JNK1/2/3, CREB, and P38α are downstream from CB1R/β-arrestin 1. During the investigation of the genes modulated by these cascades, it was discovered that of the genes specifically modulated by β-arrestins, ∼70% control gene transcription and protein synthesis, suggesting a significant role of this signaling wave in the long-term effects of CB1R activation. Remarkably, VEGFA, GH1, and ADAMTS1, genes that have been involved in cancer growth and neurodegeneration, are among the genes specifically regulated by β-arrestins (Delgado-Peraza et al., 2016).
Biased CB1Rs were also generated by mutations in the highly conserved Asp-Arg-Tyr (DRY) motif (Gyombolai et al., 2015). Either G-protein or β-arrestin 1/2–biased receptors were reported based on Go and β-arrestin 1/2 BRET responses in Chinese hamster ovary cells. Inhibition of forskolin-induced cAMP accumulation in Chinese hamster ovary cells and increased pERK levels by the AAY mutant suggested a β-arrestin bias, which supports the idea that mutations at the CB1R intracellular loop 2 comprising the β-arrestin binding site can significantly modify receptor signaling (Gyombolai et al., 2015).
Future Directions
Over the last 5 years, we have witnessed a significant increase in our knowledge of the mechanisms and signaling cascades controlled by the multifunctional scaffold protein, β-arrestin, downstream from the CB1R. In general, mounting evidence indicates that signaling cascades can be directly regulated by β-arrestin 1, whereas CB1R endocytosis is regulated by β-arrestin 2 (Ahn et al., 2013; Gyombolai et al., 2013; Srivastava et al., 2015; Delgado-Peraza et al., 2016). These signaling cascades control specific gene transcription, providing an initial glimpse to the physiologic roles of β-arrestin–mediated signaling. Recent advances on transcriptomics, signaling screenings, and computational analysis have allowed the assessment of β-arrestin–mediated signaling on a global scale. For example, β-arrestin–mediated transcriptomic signatures were shown to be conserved in vivo and in vitro, sometimes across multiple tissues, suggesting conserved biologic responses (Gesty-Palmer et al., 2013; Maudsley et al., 2015). Among them, there is a common core of biologic functions regulated by β-arrestins, such as cell growth and survival, as reported for the CB1R (Delgado-Peraza et al., 2016), and these core molecular targets could potentially mediate some of the long-term effects of CB1R activation. It is interesting to note that some of the genes specifically regulated by β-arrestins downstream from the CB1receptor control vasculature growth and prosurvival aspects of the ER-stress response, potentially explaining some of the therapeutic effects of the cannabinoid system and the effect of long-term exposure to cannabis.
Bias analysis by systematic investigation of ligands utilizing multiple readouts followed by careful quantification by bias factor analysis should provide a better understanding of the multiple biologic effects of GPCR activation and the specific effect of ligands (Stahl et al., 2015). This information should be obtained from physiologically relevant cells where receptor and signaling molecule expression levels are known and within physiologic levels to help profile novel therapeutic agents and unravel the pharmacology of the receptor (Kenakin and Christopoulos, 2013; Kenakin, 2014; Masuho et al., 2015; Maudsley et al., 2015).
Fundamental questions still remain, however. For example, why would the same cascades (e.g., ERK, CREB) be activated by different pathways (G proteins and β-arrestins) but with different spatiotemporal profiles? How do cells integrate these responses, and how conserved or relevant are these effects in vivo? Importantly, what are the physiologic roles of β-arrestin– mediated signaling, and can we specifically target these cascades for therapeutic goals? These are some of the fundamental questions the field will try to address in the coming years.
Acknowledgments
The authors offer apologies to all the authors whose work was not included in this review and thank Dr. Debra Kendall for feedback and continuous scientific discussions.
Authorship Contributions
Wrote or contributed to the writing of the manuscript:Nogueras-Ortiz, Yudowski.
Footnotes
- Received April 1, 2016.
- Accepted June 22, 2016.
-
This work was supported by the following research grants from the National Institutes of Health to G.A.Y. and C.N.O. [Grants DA023444 and R01DA037924]; G.A.Y and F.D.P. G.A.Y received further support from the Puerto Rico Science Trust.
Abbreviations
- AEA
- anandamide
- CB1R
- cannabinoid 1 receptor
- CP
- CP55,940
- CREB
- cAMP response element–binding protein
- ∆9-THC
- ∆9-tetrahydrocannabinol
- ERK1/2
- extracellular signal-regulated protein kinases 1 and 2
- GPCRs
- G protein–coupled receptors
- GRK
- G protein–coupled receptor kinases
- JNK
- c-Jun N-terminal kinase
- p38a
- mitogen-activated protein kinase 14
- PTX
- pertussis toxin
- Src
- SRC proto-oncogene, nonreceptor tyrosine kinase
- 2-AG
- 2-arachidonoylglycerol
- WIN
- WIN55,212-2
- Copyright © 2016 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license http://creativecommons.org/licenses/by-nc/4.0/.
References
