

Lumír Ondřej Hanuš *a, Stefan Martin Meyer b, Eduardo Muñoz c, Orazio Taglialatela-Scafati d and Giovanni Appendino *e
aInstitute for Drug Research, School of Pharmacy, Faculty of Medicine, Hebrew University, Ein Kerem Campus, Jerusalem 91120, Israel. E-mail: lumirh@ekmd.huji.ac.il; Fax: +972-2-6439935; Tel: +972-2-6758042
bPhytoplant Research S. L, Rabanales 21 – The Science and Technology Park of Cordoba, 14014, Cordoba, Spain
cMaimonides Biomedical Research Institute of Córdoba, Reina Sofía University Hospital, Department of Cell Biology, Physiology and Immunology, University of Córdoba, Avda Menéndez Pidal s/n., 14004, Córdoba, Spain
dDipartimento di Farmacia, Università di Napoli Federico II, Via Montesano 49, 80131 Napoli, Italy
eDipartimento di Scienze del Farmaco, Università del Piemonte Orientale, Largo Donegani 2, 28100 Novara, Italy. E-mail: giovanni.appendino@uniupo.it; Fax: +39-0321-375621; Tel: +39-0321-375621
First published on the web 10th October 2016
Covering up to January 2016
 Cannabis sativa L. is a prolific, but not exclusive, producer of a diverse group of isoprenylated resorcinyl polyketides collectively known as phytocannabinoids. The modular nature of the pathways that merge into the phytocannabinoid chemotype translates in differences in the nature of the resorcinyl side-chain and the degree of oligomerization of the isoprenyl residue, making the definition of phytocannabinoid elusive from a structural standpoint. A biogenetic definition is therefore proposed, splitting the phytocannabinoid chemotype into an alkyl- and a β-aralklyl version, and discussing the relationships between phytocannabinoids from different sources (higher plants, liverworts, fungi). The startling diversity of cannabis phytocannabinoids might be, at least in part, the result of non-enzymatic transformations induced by heat, light, and atmospheric oxygen on a limited set of major constituents (CBG, CBD, Δ9-THC and CBC and their corresponding acidic versions), whose degradation is detailed to emphasize this possibility. The diversity of metabotropic (cannabinoid receptors), ionotropic (thermos-TRPs), and transcription factors (PPARs) targeted by phytocannabinoids is discussed. The integrated inventory of these compounds and their biological macromolecular end-points highlights the opportunities that phytocannabinoids offer to access desirable drug-like space beyond the one associated to the narcotic target CB1.
Cannabis sativa L. is a prolific, but not exclusive, producer of a diverse group of isoprenylated resorcinyl polyketides collectively known as phytocannabinoids. The modular nature of the pathways that merge into the phytocannabinoid chemotype translates in differences in the nature of the resorcinyl side-chain and the degree of oligomerization of the isoprenyl residue, making the definition of phytocannabinoid elusive from a structural standpoint. A biogenetic definition is therefore proposed, splitting the phytocannabinoid chemotype into an alkyl- and a β-aralklyl version, and discussing the relationships between phytocannabinoids from different sources (higher plants, liverworts, fungi). The startling diversity of cannabis phytocannabinoids might be, at least in part, the result of non-enzymatic transformations induced by heat, light, and atmospheric oxygen on a limited set of major constituents (CBG, CBD, Δ9-THC and CBC and their corresponding acidic versions), whose degradation is detailed to emphasize this possibility. The diversity of metabotropic (cannabinoid receptors), ionotropic (thermos-TRPs), and transcription factors (PPARs) targeted by phytocannabinoids is discussed. The integrated inventory of these compounds and their biological macromolecular end-points highlights the opportunities that phytocannabinoids offer to access desirable drug-like space beyond the one associated to the narcotic target CB1.
 Lumír Ondřej Hanuš Lumír Ondřej Hanuš |
Lumír Ondřej Hanuš is an analytical chemist. He has worked for 46 years on cannabis and 26 years on endocannabinoids. He obtained his M.S. in analytical chemistry (1972) and his Ph.D. in analytical chemistry (1974) from Palacký University (Olomouc, Czech Republic), where he became associate Professor in organic chemistry in 1994. In 1995 be obtained a D.Sc. in pharmaceutical chemistry from the Charles University (Prague, Czech Republic), and he carried out reseach at Mississippi University (1978–1979), NIDDK, NIH (1997–1998), and NIAAA, NIH (2001–2002). In 1992, he and molecular pharmacologist William Devane discovered the first endocannabinoid (anandamide). He is currently a research fellow at the Hebrew University in Jerusalem, and has been awarded, among others honors, a Chemiae Doctor honoris causa (2007) and Medicinae Doctor honoris causa (2011). |
 Stefan Martin Meyer Stefan Martin Meyer |
Stefan Martin Meyer, MBA, is CEO of Phytoplant Research S.L. and President of the BoD of Vivacell Biotechnology España S.L, two companies involved in phytocannabinoid research. He has a decade of experience in the development of medicinal and healthfood cannabis-based products. |
 Eduardo Muñoz Eduardo Muñoz |
Eduardo Muñoz is Professor of Immunology at the Department of Cell Biology, Physiology and Immunology at the University of Córdoba (Spain). His research activity is centered on the anticancer and antiinflammatory activities of endocannabinoids and novel cannabinoid derivatives as well as other natural products. He is the author of more than 160 papers in scientific journals and patents. He is a member of the Editorial Board of Planta Medica and Fitoterapia. |
 Orazio Taglialatela-Scafati Orazio Taglialatela-Scafati |
Orazio Taglialatela-Scafati is Professor of Pharmaceutical Biology at the Department of Pharmacy, University of Naples Federico II. His scientific interests include isolation, stereostructural characterization, and modification of secondary metabolites from marine invertebrates and terrestrial plants, to be used as leads in drug discovery or as tools to investigate biology. He is the author of more than 140 papers in scientific journals and has edited the books “Flavour and Fragrance Chemistry”, “Modern Alkaloids” and “Handbook of Marine Natural Products”. He is an Associate Editor of Marine Drugs and a member of the Editorial Boards of Acta Pharmaceutica Sinica, Steroids and Fitoterapia. |
 Giovanni Appendino Giovanni Appendino |
Giovanni Appendino is Professor of Chemistry at the Università del Piemonte Orientale, Department of Pharmaceutical Sciences, Novara (Italy). His research activity is centered on the isolation, chemical modification, and total synthesis of bioactive natural products of plant origin. In recognition of his studies on bioactive natural products, he received the Rhône-Poulenc Rorer Award of the Phytochemical Society of Europe in 1991, the Medaglia Quilico of the Società Chimica Italiana in 2009, and the Bruker Prize of the Phytochemical Society of Europe in 2014. He is author of over 350 journal articles and 15 book chapters. He is editor-in-chief of the Journal Fitoterapia, and a member of the advisory board of Natural Product Reports, Progress in the Chemistry of Organic Natural Products, Phytochemistry Letters, Natural Products Communications and Acta Pharmaceutica Sinica. |
1. Introduction
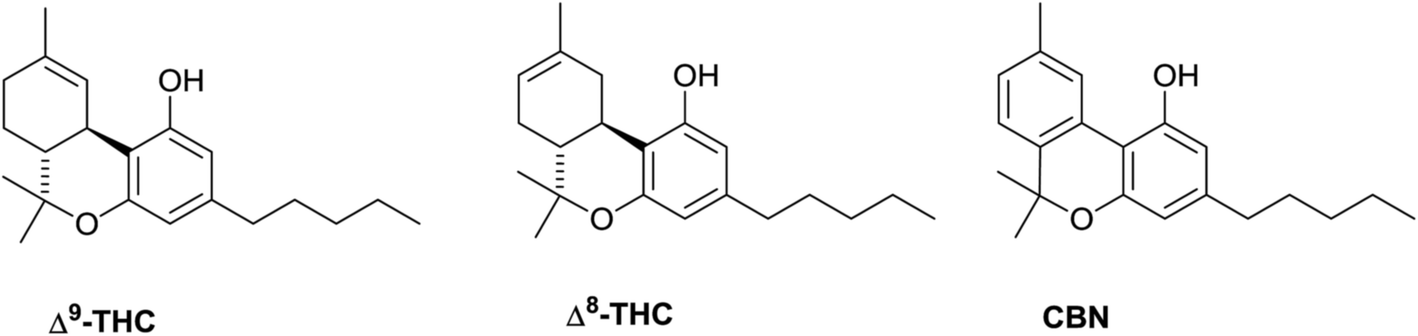
![]() Over the past decades, the name “cannabinoid” has become increasingly vague. Originally coined in a phytochemical context to refer to a structurally homogenous class of meroterpenoids typical of cannabis (Cannabis sativa L.), the name “cannabinoid” has then been associated to the biological profile of the psychotropic constituent of marijuana (Δ9-THC), substantially losing its structural meaning and being growingly associated, in accordance with the rules of pharmacological research,1 to compounds showing affinity to the two GPCR known as cannabinoid receptors (CB1 and CB2), independently from any structural or biogenetic relationship with the cannabis meroterpenoids. To compound semantics even more, CB1 and CB2 are actually Δ9-THC receptors, since, within the almost 200 known cannabinoids, only Δ9-THC, its isomer Δ8-THC, and, to a lower extent, their aromatized derivative CBN (Fig. 1), bind with significant affinity the ligand recognizing site of these receptors.1 The endogenously produced biological analogues of THC are referred to as endocannabinoids,1 and it seems therefore logical to refer to cannabis meroterpenoids and their analogues of plant origin as phytocannabinoids, emphasizing their botanical origin.
Over the past decades, the name “cannabinoid” has become increasingly vague. Originally coined in a phytochemical context to refer to a structurally homogenous class of meroterpenoids typical of cannabis (Cannabis sativa L.), the name “cannabinoid” has then been associated to the biological profile of the psychotropic constituent of marijuana (Δ9-THC), substantially losing its structural meaning and being growingly associated, in accordance with the rules of pharmacological research,1 to compounds showing affinity to the two GPCR known as cannabinoid receptors (CB1 and CB2), independently from any structural or biogenetic relationship with the cannabis meroterpenoids. To compound semantics even more, CB1 and CB2 are actually Δ9-THC receptors, since, within the almost 200 known cannabinoids, only Δ9-THC, its isomer Δ8-THC, and, to a lower extent, their aromatized derivative CBN (Fig. 1), bind with significant affinity the ligand recognizing site of these receptors.1 The endogenously produced biological analogues of THC are referred to as endocannabinoids,1 and it seems therefore logical to refer to cannabis meroterpenoids and their analogues of plant origin as phytocannabinoids, emphasizing their botanical origin.
 |
||
| Fig. 1 High-affinity phytocannabinoid ligands of cannabinoid receptors. | ||
The phytocannabinoid structural motif is biogenetically hybrid, and results from the convergence of the mevalonate and the polyketide pathways. Since both of them are intrinsically modular, variation in terms of polyketide starter and prenyl oligomerization are possible, and indeed Nature has deftly capitalized on this modularity to create chemical diversity that complements the one resulting from the oxidative cyclase phase of isoprenyl diversification. As a result, the name phytocannabinoid is also vague from a structural standpoint. The biogenetic hallmark of phytocannabinoids is a resorcinyl core decorated with para-oriented terpenyl and pentyl groups, but compounds with a different degree of isoprenylation (prenyl, sesquiterpenyl) or with a shortened alkyl group (methyl, propyl, or more rarely ethyl and butyl) are also present in C. sativa. Phytocannabinoids derived from aliphatic ketide starters are typical of C. sativa and are otherwise of limited distribution in Nature, while their analogues derived from an aromatic ketide starter and with a phenetyl-type substituent have a much broader distribution, encompassing not only plants but also liverworts and fungi. Many of these compounds are referred to in the literature as prenylated bibenzyls, a name that hides their relationship with their more famous analogues from cannabis.
To cope with the biogenetic abundance associated with the production of cannabinoids, we propose the classification summarized in Table 1 to address variation of the substituents of the resorcinyl core and of their topological relationships. According to this proposal, “classic” phytocannabinoids are those whose resorcinyl side-chain is derived from a linear aliphatic polyketide starter, while their analogues derived from aromatic starters could be referred to as aralkyl phytocannabinoids. Regarding the relationship between the substituents of the resorcinyl moiety, in most compounds isoprenyl and the resorcinyl side-chain are para-related, while analogues where these groups are in an ortho-relationship are assigned to the “abnormal” series. Finally, compounds characterized by an elongated or a shortened terpenyl residue should be referred to as sesquicannabinoids when the isoprenyl residue is of the sesquiterpenyl type, and deprenylcannabinoids when the isoprenyl residue is a simple dimethylallyl. Most cannabinoids have so far been isolated as artifacts from their carboxylated forms (pre-cannabinoids or acidic cannabinoids) from plant sources, and are therefore phytocannabinoids, but the generality of the biogenetic origin does not make it unconceivable that compounds of this type could also occur in fungi or bacteria, and some examples of fungal cannabinoids are indeed known. While phytocannabinoids from the abnormal- and the sesquiterpenyl-series occur in cannabis, phytocannabinoids derived from an aromatic ketide starter have never been reported from this plant source.
| Compound class | Ketide starter | Side-chain/isoprenyl topological relationship | Isoprenyl residue |
|---|---|---|---|
| Alkyl phytocannabinoids | Aliphatic | para | Terpenyl (C10)-type |
| Aralkyl phytocannabinoids | Aromatic | para | Terpenyl (C10)-type |
| Abnormal series | Aliphatic or aromatic | ortho | Isoprenyl |
| Sesqui (deprenyl)-series | Aliphatic or aromatic | ortho or para | Sesquiterpenyl (C15) |
| Deprenyl (C5) |
This review article aims at providing a comprehensive inventory of phytocannabinoids of different botanical origin. Most phytocannabinoids chemotypes were characterized in the 60ties and 70ties,2–5 but, after a three-decade gap, new structural types have been discovered, as exemplified by sesquicannabinoids6 and by the isoprenyl esters of pre-cannabinoids.7 Furthermore, technological advancement, the growth of the natural product community, and the availability of new cannabis breeds are expected to further expand the current inventory of these compounds. Most phytochemical studies on cannabis precede the identification of cannabinoid and TRPs receptors that occurred in the 90ties, and bioactivity was mostly evaluated with the cannabinoid tetrad test in mice, a combination of four different behavioural tests (hypothermia, hypomotility, catalepsy, analgesia) that, although per se unspecific, when all four positive were indicative of a Δ9-THC-type activity.8 Activities unrelated to the activation of CB1 and the replication of the biological profile of Δ9-THC were therefore missed.
Various articles have regularly updated the inventory of phytocannabinoids from C. sativa,2–5 but no attempt has so far been done to include in this survey also phytocannabinoids from additional natural sources. Apart from this, we have also tried to outline the basic chemical and biological profile of the various structural types of phytocannabinoids, and to discuss their biogenetic relationships, chemical interconversions, and biomimetic synthesis from terpene derivatives and resorcinols.
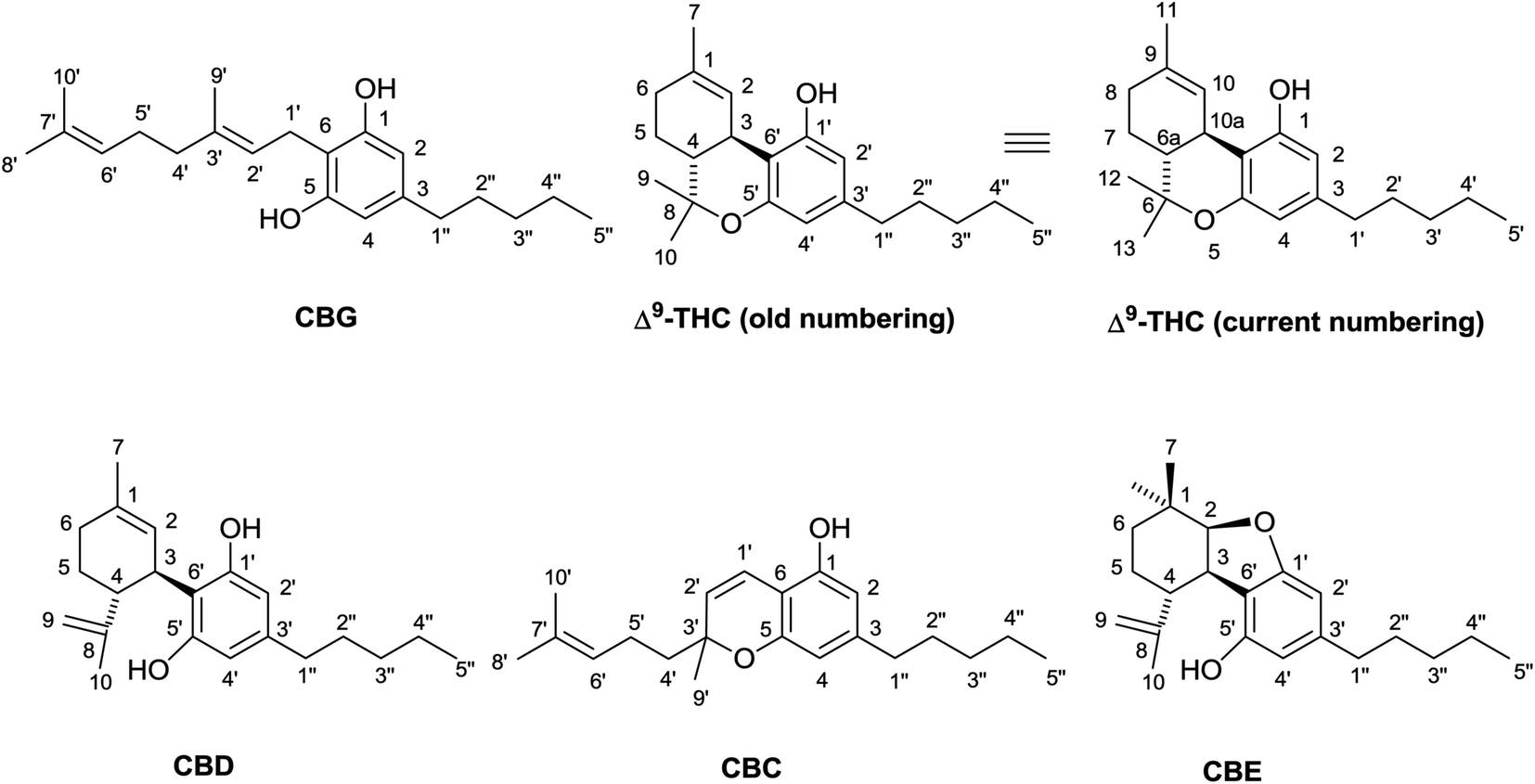
The most important phytocannabinoids are commonly referred to using a three-letter acronym system originating from the first investigators in the field, and later updated by ElSohly to include all the major structural types (Fig. 2).5 Regrettably, there is no single numbering throughout the various classes of phytocannabinoids, and at least five different systems are documented in the literature. As a rule, the reference system is given simple numbers, while positions in the other elements are referred to with primed or doubly primed numbers. There is no agreement, however, on the identification of the reference system. It used to be the terpene moiety in all cases, but it is now growingly considered the aromatic ring in CBG derivatives (but not in CBD). When oxygen bridges are present between the terpenyl and the resorcinyl system, the reference system becomes the corresponding fused heterocycle in accordance with the IUPAC rules, even though this hides relationships between biogenetically corresponding carbons (Fig. 2). Thus, all p-menthane-type phytocannabinoids were originally numbered in the same way, using the isoprenoid moiety as a basic system, but, also because of ambiguities in the identification of the starting carbon of the menthane moiety (benzylic carbon vs. the methyl-bearing olefin carbon), the terpenoid numbering has now been replaced by the heterocyclic numbering. As a result of this change, Δ9-tetrahydrocannabinol (Δ9-THC) and cannabidiol (CBD), although structurally related (Scheme 1), are numbered in a different way (Fig. 2). The terpenoid system is still often used for cannabichromene (CBC) and for cannabicyclol (CBL), both numbered according to CBG, while cannabielsoin (CBE) is numbered according to THC. To avoid confusion, especially when tabulating NMR data, it would be practical to have a reference numbering system capable to accommodate all phytocannabinoids having the same type of isoprenyl residue, independently from the closure of oxygenated heterocyclic with the resorcinyl moiety.
 |
||
| Fig. 2 Phytocannabinoid numbering systems. | ||
 |
||
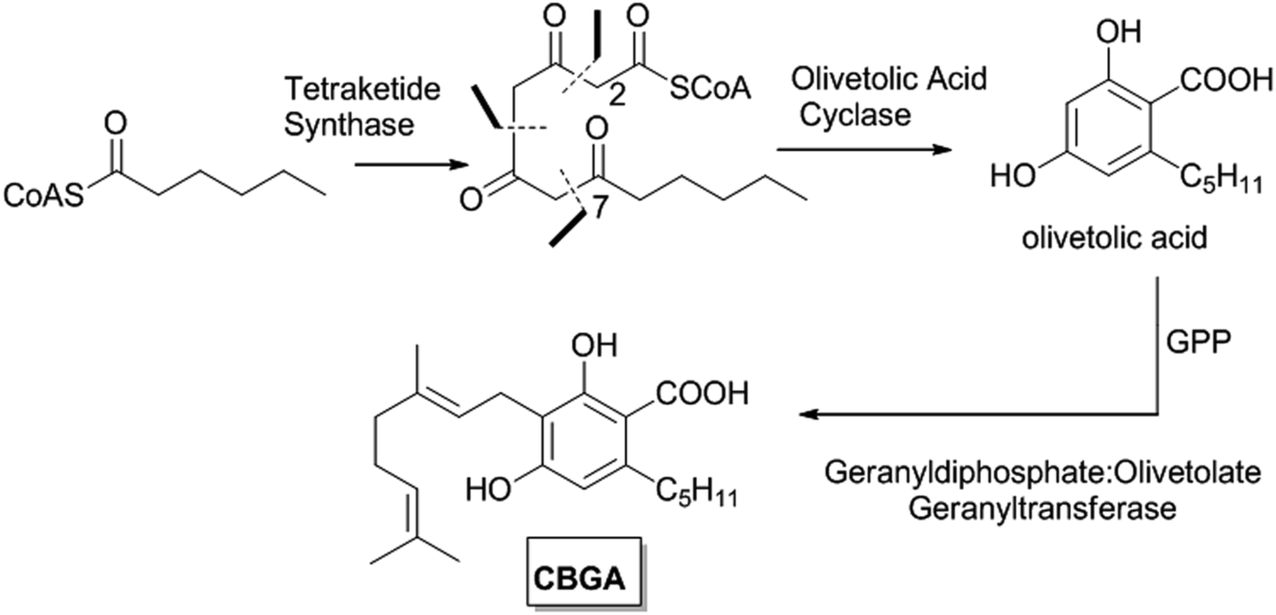
| Scheme 1 Formation of cannabigerolic acid (CBGA) in C. sativa. | ||
2. Biogenesis of phytocannabinoids
Neutral phytocannabinoids were long assumed to be genuine natural products, but, while investigating fresh samples of fiber hemp, Schulz and Haffner9 discovered that their major constituent was not CB, but, rather, its carboxylated version (cannabidiolic acid, CBDA or pre-CBD, Scheme 2), a compound first described by Krejčí and Šantavý in 1955.10 It is currently assumed that all neutral phytocannabinoids originate from the mostly non-enzymatic decarboxylation of their corresponding carboxylated forms. Consequently, olivetolic acid and not olivetol, was their actual aromatic precursor, and the early biogenetic schemes were elaborated on the basis of the biosynthesis of polyketides, identifying some basic relationships between the small pool of the compounds known at that time. The first step in cannabinoid biosynthesis was correctly considered the condensation of a hexanoylCoA and three activated acetate units to generate the diketo tautomer of olivetolic acid. Farmilo’s biogenetic proposal11 was the first to consider phytocannabinoids in their native carboxylated form, anticipating the existence of THCA before its actual isolation.
 |
||
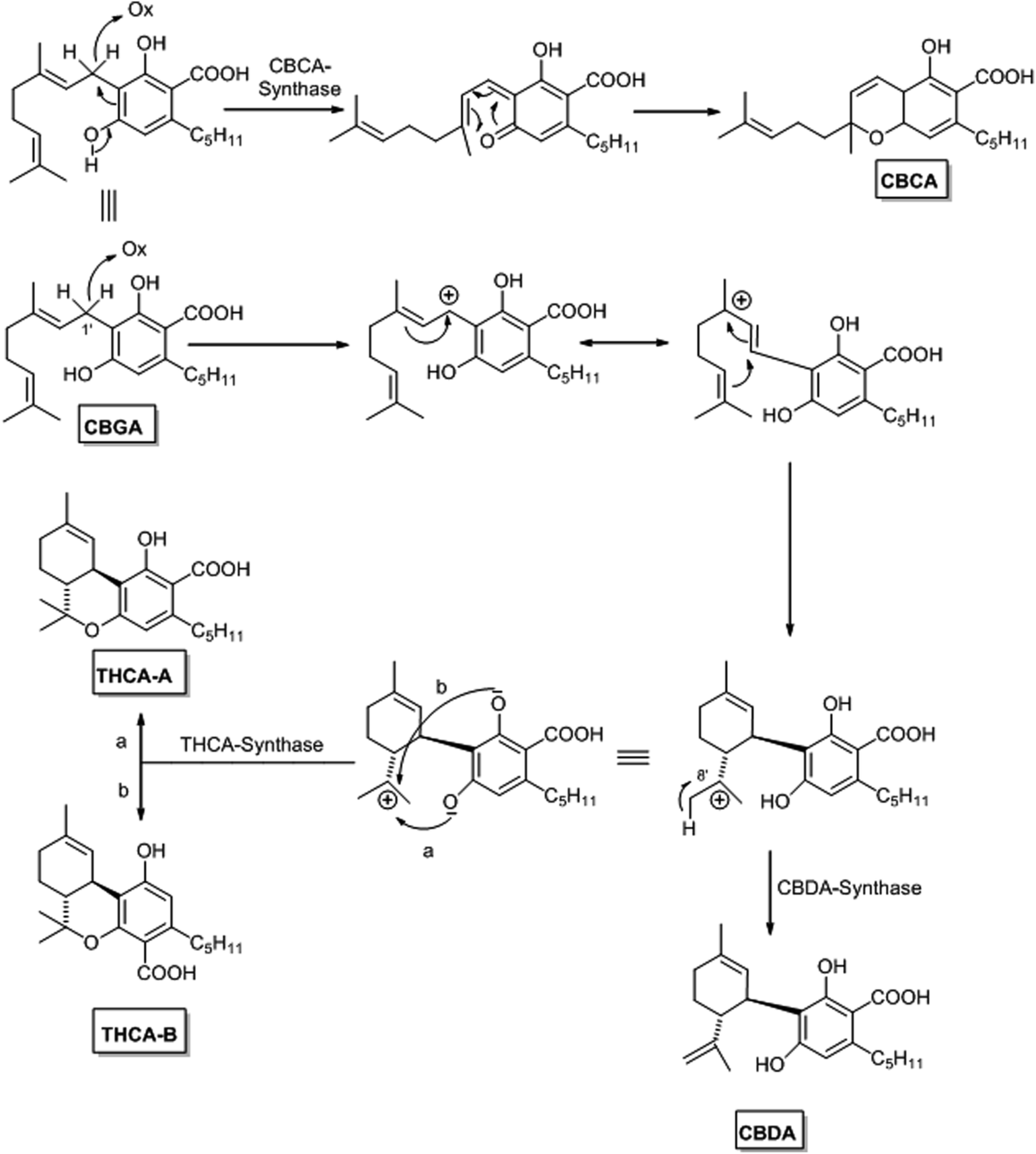
| Scheme 2 Biosynthetic origin of the major phytocannabinoids. | ||
Guided by this proposal, the enzymology of phytocannabinoids biosynthesis was substantially clarified. A polyketide origin for the resorcinyl moiety of phytocannabinoids is consistent with the finding that a close relationship exists in Cannabis tissues (female flowering tops, leaves, stems and roots) between the levels of hexanoylCoA and the concentrations of the carboxylated form of CBD (pre-CBD, CBDA). A gene encoding a novel type III polyketide synthase (PKS) was cloned from C. sativa and named olivetol synthase,12 but the enzyme actually failed to produce olivetol or olivetolic acid in the absence of a polyketide cyclase enzyme, named olivetolic acid cyclase (OAC) that was cloned from the glandular trichomes of cannabis.13 This enzyme catalyzes a C-2/C-7 intramolecular aldol condensation, retaining the carboxylic group and forming olivetolic acid. Interestingly, OAC is a dimeric α + β barrel (DABB) protein structurally similar to polyketide cyclases from Streptomyces species, indicating evolutionary parallels between polyketide biosynthesis in plants and bacteria.14
Regarding the isoprenoid residue, Mechoulam recognized CBG as the precursors of all other types of phytocannabinoids already in 1964,15 reasoning that this compound has the lowest oxidation level for the isoprenyl moiety. Accordingly, CBG can be formed by the C-isoprenylation of olivetolic acid with geranyl diphosphate, and then be converted to CBD, THC and, eventually, CBN. Two years later, the biogenesis of cannabinoids from geranyldiphosphate and olivetolic acid was indeed reported.16 This biogenetic bluepring was next extended17 to include the possibility to generate both acidic and neutral cannabinoids, with, however, growing awareness that neutral phytocannabinoids might actually be artifacts formed during harvest and storage of Cannabis.18
Progress was done in the discovery of the enzymes responsible for the isoprenylation of olivetolic acid, and a specific enzyme, named geranyldiphosphate:olivetolate geranyltransferase, was characterized in young leaves of C. sativa.19 This enzyme catalyzes the first step in cannabinoid formation in hemp, namely the prenylation of olivetolic acid, and accepts geranyldiphosphate (in turn derived from the plastidial 2-methyl-D-erythritol-4-phosphate pathway) as a substrate. In the presence of olivetolic acid (olivetol is not accepted as a substrate), a ca. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cannabigerolic- and cannabinerolic acids is formed. The replacement of geranyldiphosphate with neryldiphosphate changed the ratio to 1:1, with rate being only 20% of the one observed with geranyldiphosphate.19
:1 mixture of cannabigerolic- and cannabinerolic acids is formed. The replacement of geranyldiphosphate with neryldiphosphate changed the ratio to 1:1, with rate being only 20% of the one observed with geranyldiphosphate.19
The isoprenylation step is next followed by an oxidative cyclase activity that, through the agency of specific enzymes, generates CBCA, CBDA and Δ9-THCA from CBGA. From a mechanistic standpoint (Scheme 2), the reaction formally involves hydride abstraction from the benzallylic terpenyl carbon. The formation of the resulting cation scrambles the configuration of the adjacent double bond, making it possible the generation of the cyclohexene ring of CBDA and Δ9-THCA by electrophilic cyclization. Alternatively, the isomerized benzallyl cation can evolve into a quinone methide and generate CBCA by an electrocyclic reaction. The electrophilic cyclization is enzyme-promoted and generates chiral products, while the electrocyclic reaction is probably spontaneous, since CBCA is generated as a racemate.
The electrophilic cyclization step is highly specific in terms of termination. In one version of the process, the C-8 cation (menthane numbering) behaves as a Broensted acid, and is quenched by loss of a proton from C-9 to generate the exocyclic double bond of CBDA (Scheme 2). In the alternative version of the termination, the C-8 menthyl cation behaves as an electrophilic sink for one of the two ortho-hydroxyls, generating Δ9-THCA-A from the hydroxyl para– to the carboxylate, and Δ9-THCA-B from the other phenolic hydroxyl. The oxidative- and the electrophilic cyclase activities are closely associated, and the menthyl cation is not released or leaking from the enzymatic cleft where it is generated, making the two termination process biogenetically orthogonal. This is consistent with the paradoxical observation that, while CBD is easily converted into Δ8– and Δ9-THC by acidic treatment under laboratory conditions, CBDA is not converted into THCA in cannabis tissues. CBDA-synthase and THCA-synthase have been cloned from the storage cavity of the glandular trichomes of cannabis,20,21 and they exclusively produced their corresponding phytocannabinoids. THCA synthase has also been crystallized, and the FAD and substrate-binding sites identified.22 Apparently, the enzyme selectively produce one of the two isomeric THC acids present in nature, THCA-A.22 THCA- and CBDA-synthases are similar in terms of mass (both are 74 kDa monomeric proteins), pI, vmax and Km for CBGA, and are 84% identical in their amino-acid sequence.21,23 Both THCA- and CBDA-synthases show a domain with high homology with the enzyme involved in the oxidative cyclization step of the biosynthesis of berberine, a benzophenantridine alkaloid, in the Californian poppy (Eschscholtzia californica). Both processes require molecular oxygen for their activity and form hydrogen peroxide during the oxidative cyclization of the substrate.24 Also cannabichromenic acid (CBCA) synthase, the enzyme catalyzing the oxidocyclization of CBGA to CBCA has been identified in young leaves of cannabis and next purified and characterized.25 A summary of the biogenic relationship between the main phytocannabinoids in Cannabis sativa L. is reported in Scheme 2.
Genuine oxidative capacity has been detected in cannabis tissues, as shown by the observation that suspension cultures of the plant can convert primary and secondary allylic alcohols into the corresponding carbonyls.26 It is unclear, however, whether phytocannabinoids are substrates for this activity.
Labelling experiments with 14C-CBG, and 14C-olivetolic acid were used to study the production of phytocannabinoids in cannabis roots. These experiments confirmed that C-3 phytocannabinoids derive from an independent biosynthesis and not from the enzymatic shortening of the C-5 side chain by either plant or contaminating fungal tissus.27 Thus, all the CBGA alkyl-homologs could be used as substrate for the different cannabinoid synthases in vitro, although the efficiency of conversion was different within the various homologues.27 It was also shown that decarboxylation of cannabinoid acids is a continuous process, generating neutral cannabinoids already in the early stages of the plant growth, and next continuing during all the vegetation stage.27
There is currently great interest in the expression of the key enzymes involved in the production of phytocannabinoids in fermentable organisms, and in 2015 it was announced that the methylotropic yeast Pichia pastoris has been engineered to produce Δ9-THCA from CBGA.28 Functional expression of Δ9-tetrahydrocannabinolic acid synthase (THCAS) was also obtained in baker’s yeast (Saccharomyces cerevisiae), although an overall lower fermentation yield was obtained.28
The genetic of inheritance of the enzymes responsible for the formation of the major cannabinoids is complex, and has been extensively investigated as regards CBDA and THCA synthases. These two enzymes are assumed to be coded for by two co-dominant alleles, respectively BD and BT, while a defective form of the allele could be responsible for the accumulation of CBG via the production of an inactive or minimally active oxydocyclizing enzyme. The situation is, however, complicated by the presence of a host of THCA- and CBDA-synthase-related pseudogenes that make the inheritance of phytocannabinoids substantially deviating from a simple Mendelian model.29
Nature is a biogenetically tinkerer, and prefers to re-use, recycle and re-assemble rather than creating ex novo something new. This so called “law of stinginess” is exemplified by the observation that certain isoprenylated ketides replicate, within the framework of compounds derived from an aromatic starter, the features of phytocannabinoids from cannabis (aldol-type derivation of the prenylated aromatic moiety, resorcinyl-type hydroxylation pattern, C-monoprenylation), fully qualifying as “phytocannabinoids”, as will be discussed in Section 4.2 to highlight the difference between phytocannabinoids and phytocannabinoid-like compounds.
3. Naturally occurring phytocannabinoids
3.1 Structural diversity
The diversity of natural phytocannabinoids is the result of differences in their three moieties, namely the isoprenyl residue, the resorcinyl core, and the side-chain. These differences are generally orthogonal, that is, biogenetically unrelated. Although impressive, the inventory of alkyl-cannabinoids might have been inflated by the poor oxidative stability of some of the major phytocannabinoids, Δ9-THC in particular. Furthermore, many investigations were carried out on aged samples of seized marijuana or hashish, and some compounds were only observed as GC peak and tentatively identified by their mass spectrum, without never actually have been isolated.
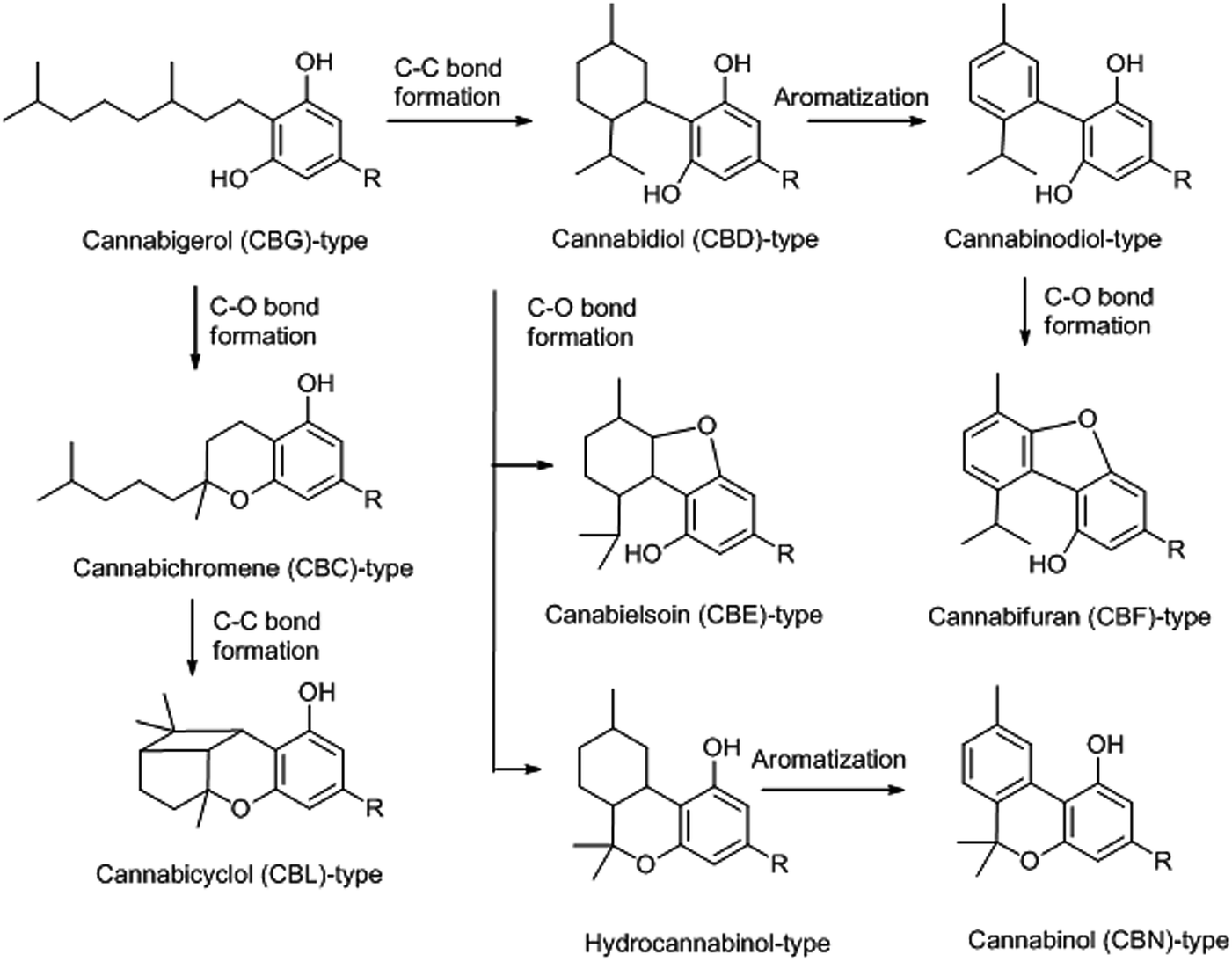 |
||
| Scheme 3 Topological classification of the major skeletal types of phytocannabinoids. | ||
(a) The carbon–carbon connectivity of their isoprenyl moiety, that can be linear (cannabigerol-type compounds), monocyclic (para-menthane-type and thymyl-type) or bicyclic (cannabicyclol-type phytocannabinoids)
(b) The closure of oxygen bridges between the isoprenyl and the resorcinyl moieties, that generates cannabichromene (CBC)-type compounds from linear precursors and hydrocannabinol-, cannabielsoin (CBE)- and cannabifuran (CBF)-type compounds from monocyclic precursors.
(c) The aromatization of the p-menthyl moiety to a thymyl moiety, that generates cannabinol-type and cannabinodiol-type derivatives from, respectively, THC- and CBD-type precursors.
(d) The closure of additional carbon-bonds, as exemplified by cannabicyclol derivatives.
Acidic cannabinoids have been detected in historical samples of Cannabis tincture over 100 year old,31 and these compounds are not decarboxylated under physiological conditions.32 Up ca. 70% decarboxylation has been reported in controlled smoking experiments,32 but the half-life of acidic phytocannabinoids in plant material at room- or lower temperatures is in the range of hundreds of days.32 Therefore these compounds are the major form of phytocannabinoids present in edible marijuana. Despite their low volatility, pre-cannabinoids are absorbed from smoked cannabis, and the detection of pre-THC derivatives has even been proposed as a diagnostic test to distinguish the recreational use of marijuana, that contains pre-THC, from positivity due to the assumption of mainstream medications originating from semi-synthetic THC (Marinol®).32 There is currently great interest for pre-cannabinoids, fostered by the discovery that pre-THC retains activity at both CB1 and CB2, but is not narcotic due to its very poor brain penetration.33 Pre-cannabinoids can also occur as thermally-stable complex esters with terpenic and sesquiterpenic alcohols, and the pharmacology of these compounds is still unexplored, probably because of the difficulty to purify them from the highly lipophic fractions of cannabis extracts. Methyl esters of pre-cannabinoids were often prepared to facilitate their purification, but hydrolysis by basic treatment to regenerate the native acids has been reported to be unsuccessful.34 Pre-cannabinoids show strong anti-bacterial activity, similar to the one of their corresponding neutral derivatives.35 Further structural diversity in the resorcinyl moiety can involve O-alkylation, generally with a methyl group, or oxidation to the quinol and hydroquinol level. Cannabinoids from the quinol series are intensively purple-colored in non-acidic conditions, and their easy formation from CBD and CBG is at the basis of the Beam test, a forensic identification method for marijuana.36 Cannabinoid quinols are unstable toward dimerization and further degradation,36 and have so far been isolated only in traces from the abnormal series,37 as their stable acetates from the normal series,36 or in deoxygenated form.38 They might also be involved in the mammalian metabolism of phytocannabinoids, but their instability and the lack of reference compounds have combined to leave this issue unsettled.39 Cannabinoid quinols show interesting bioactivity, and those derived from CBD (HU-313)40 and CBG (VCE-003)41 (Fig. 3) are non-adipogenic PPARγ agonists and have been considered for clinical development respectively, as anticancer agent and as neuroprotectory agents.40–42 These compounds could be stabilized as rapidly re-oxidized aza-Michael adducts without loss of anti-fibrotic activity as in VCE-004-8 (Fig. 3).43
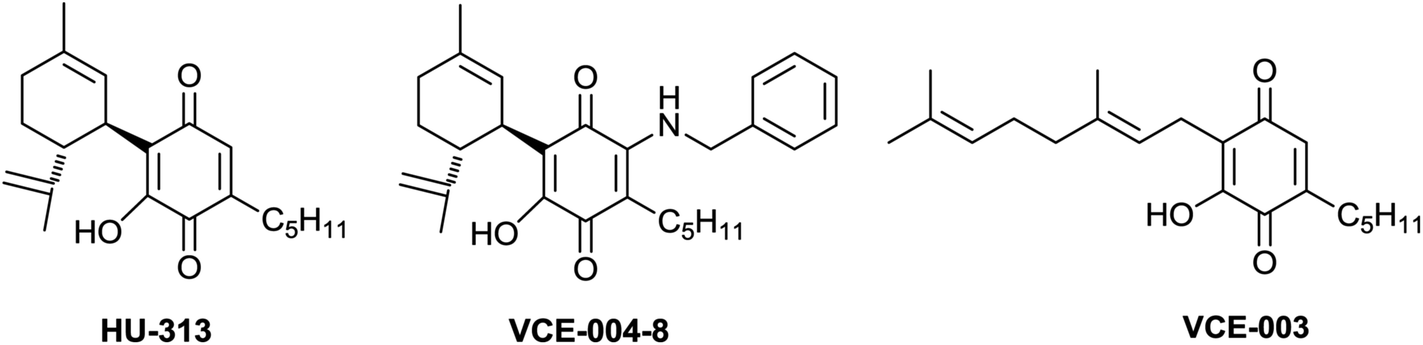 |
||
| Fig. 3 Bioactive cannabinoid quinols under preclinical/clinical development. | ||
The carbon-substitution pattern of the resorcinyl core is generally 1,4, with the isoprenyl and the side-chain para-related. Few alkyl phytocannabinoids belong to the so called “abnormal series”, where the two carbon substituents are in an ortho-relationship (Fig. 2), but these compounds are more common in aralkyl phytocannabinoids. Compounds from the abnormal series derive by a process of prenylation at the carbon in ortho or para relationship to the resorcinyl hydroxyls, while cannabinoids from the normal series derive from the alkylation of the carbon adjacent to the two resorcinyl hydroxyls (Fig. 4).
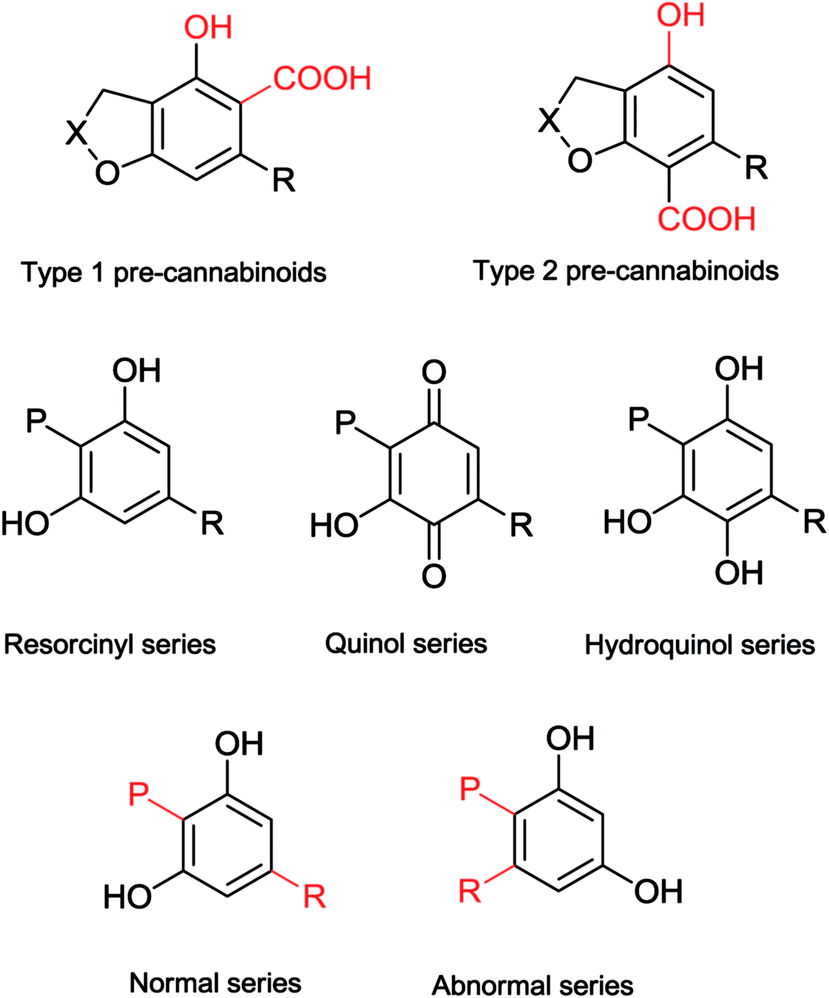 |
||
| Fig. 4 Diversity of the resorcinyl moiety of cannabinoids (P = prenyl; R = alkyl). | ||
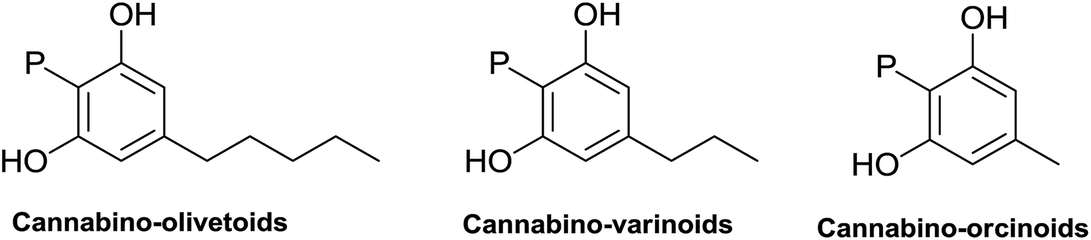 |
||
| Fig. 5 Major classes of alkyl phytocannabinoids. | ||
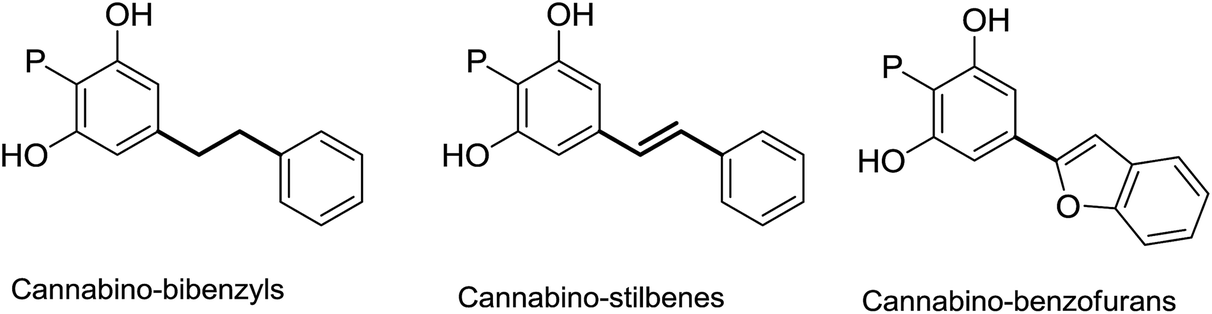
Aralkyl phytocannabinoids do not occur in Cannabis, but have an otherwise broad distribution in plants, both higher (Helichrysum, Amorpha, Glycyrrhiza and other genera) and lower (liverworts from the Radula species), with even a single report from a parasitic fungus. The aralkyl residue can be of the phenethyl-, stiryl-, or benzofuranyl type (Fig. 6), and the corresponding compounds have been named bibenzyl-, stilbenyl- and benzofuranyl phytocannabinoids.
 |
||
| Fig. 6 Major classes of aralkyl phytocannabinoids. | ||
4. Phytocannabinoids inventory
Depending on the nature of the resorcinyl side-chain, compounds will be sorted out in alkyl- and β-aralkyl phytocannabinoids. Within the two classes, compounds are classified according to the nature of the isoprenyl residue (linear, carbo-monocyclic) and the presence of oxygen bridges with the resorcinyl core, making reference to a set of archetypal major chemotypes.
4.1 Alkyl phytocannabinoids
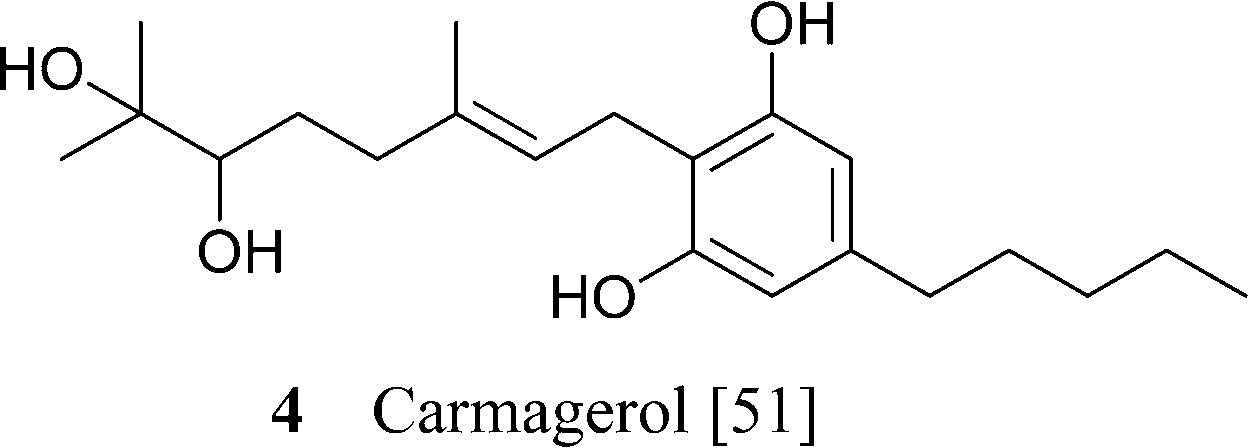
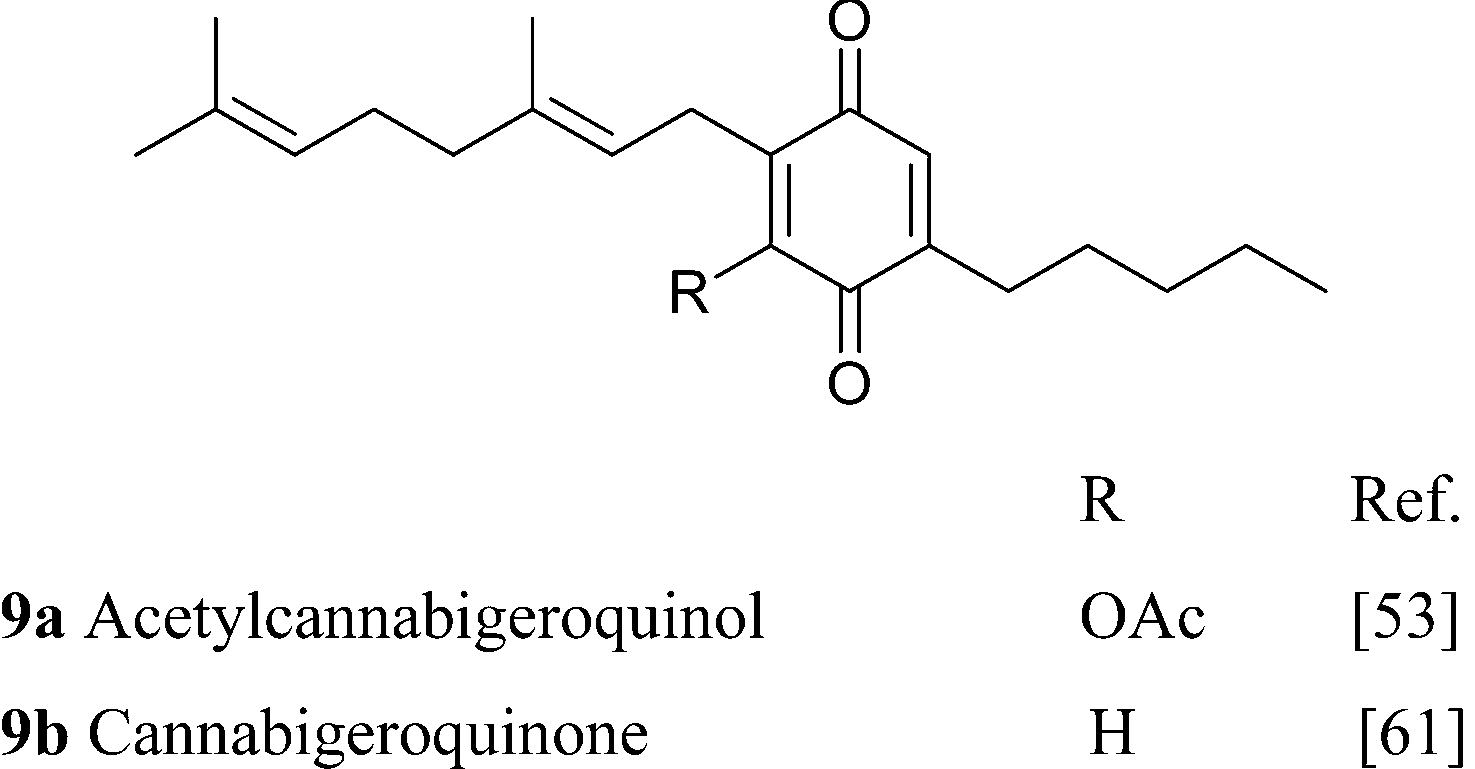
Apart from the parent compounds (CBG and CBGV) and their carboxylic forms, all the other derivatives are minor or trace constituents of cannabis, with the exception of the mono-methyl ether of CBG (1e), that occurs in significant concentrations in some Asian strain of Cannabis.50 Dihydroxylation of CBG affords chemoselectively the ω-epoxide, identical to the racemic compound (carmagerol, 4), isolated from the Carmagnola variety of fiber hemp.51 Also the proximal epoxides were isolated as a racemic mixture, from both the geranyl (CBG) and the neryl (cannabinerolic) series of neutral and acidic cannabinoids.52 Analogues with an oxidized resorcinyl residue have also been characterized, both in the quinol and the hydroxyhydroquinone form. Quinol cannabinoids are very unstable,36 and the isolation of 9a is undoubtedly due to the acetylation of one of the hydroxyl.53
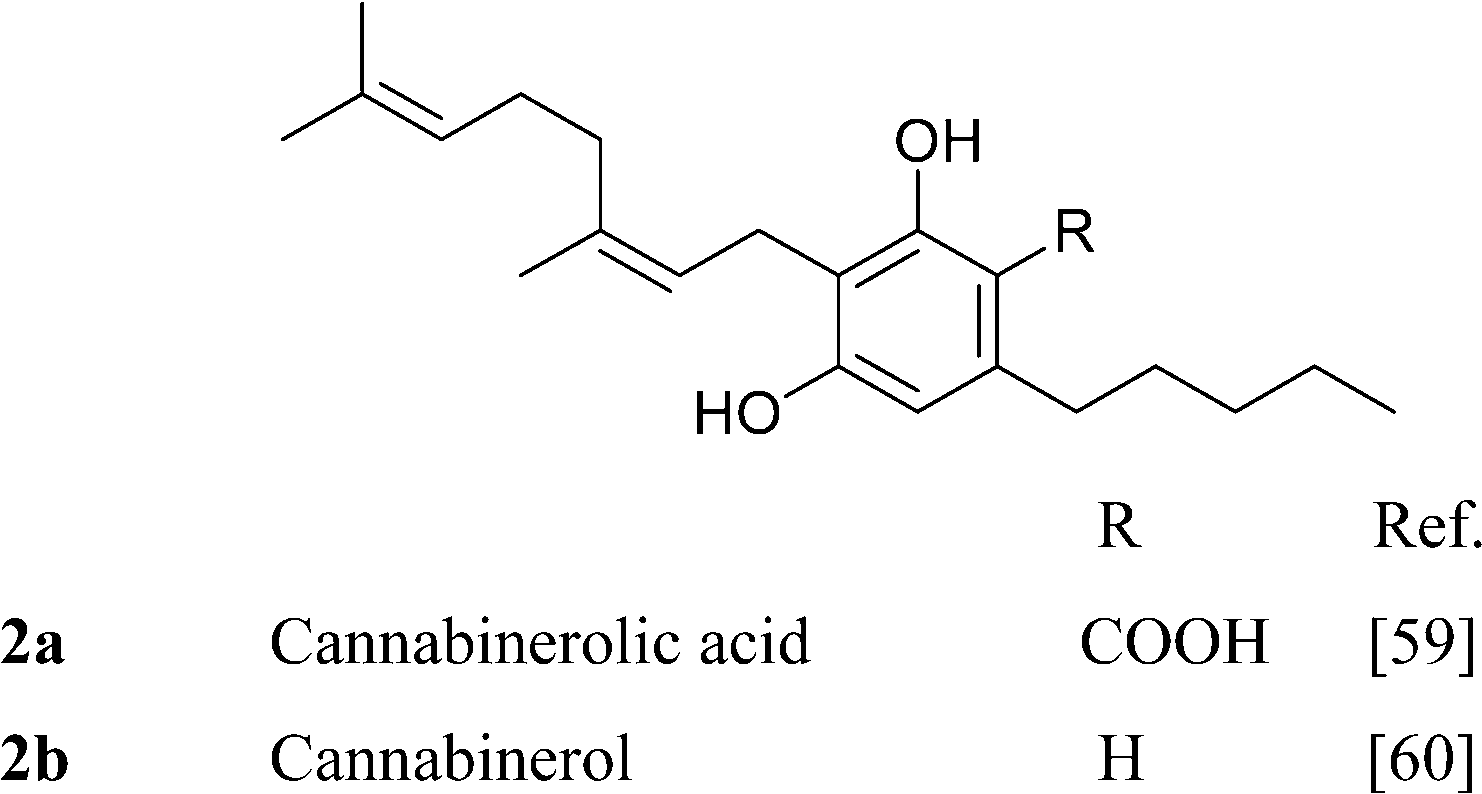
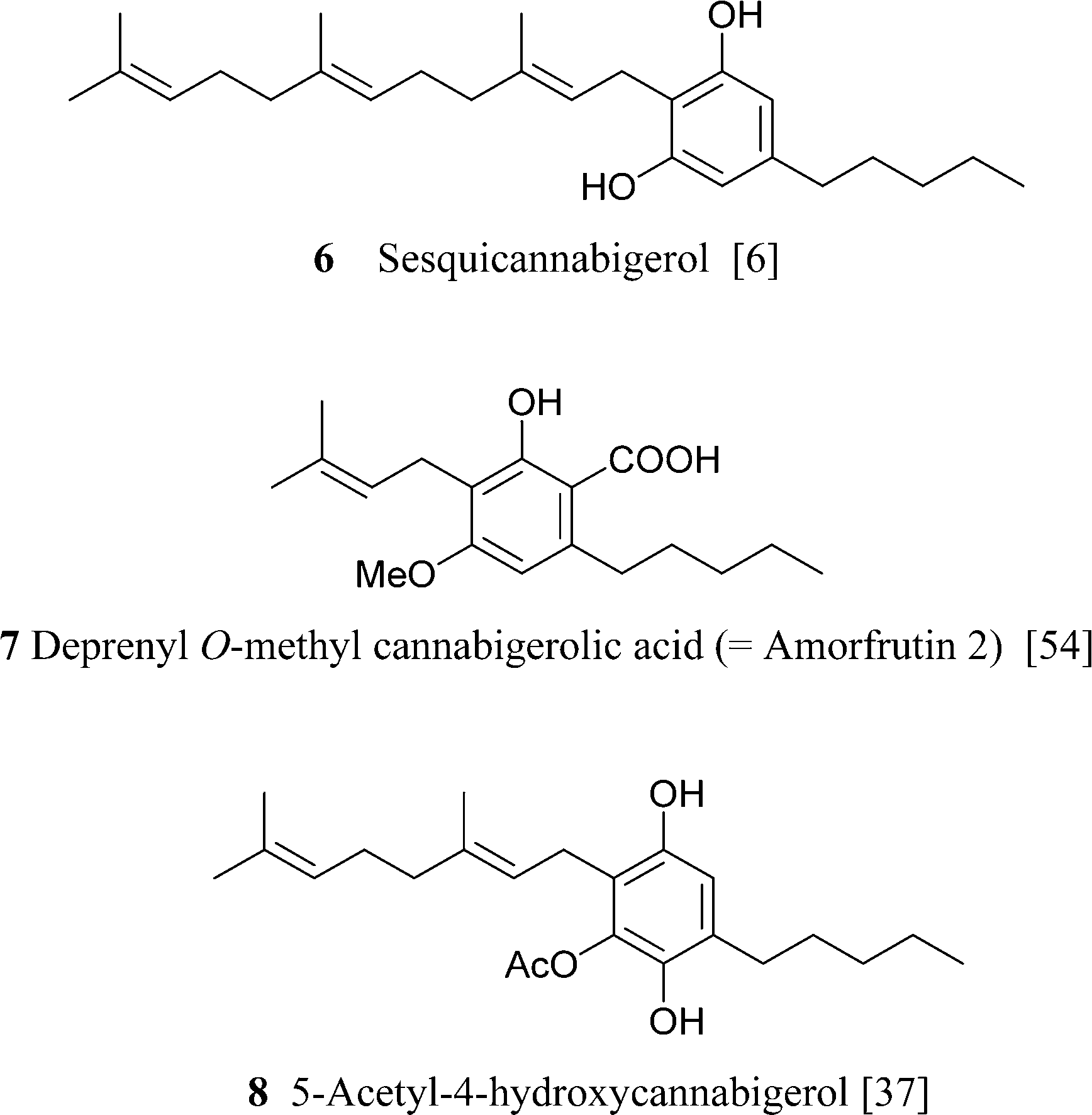
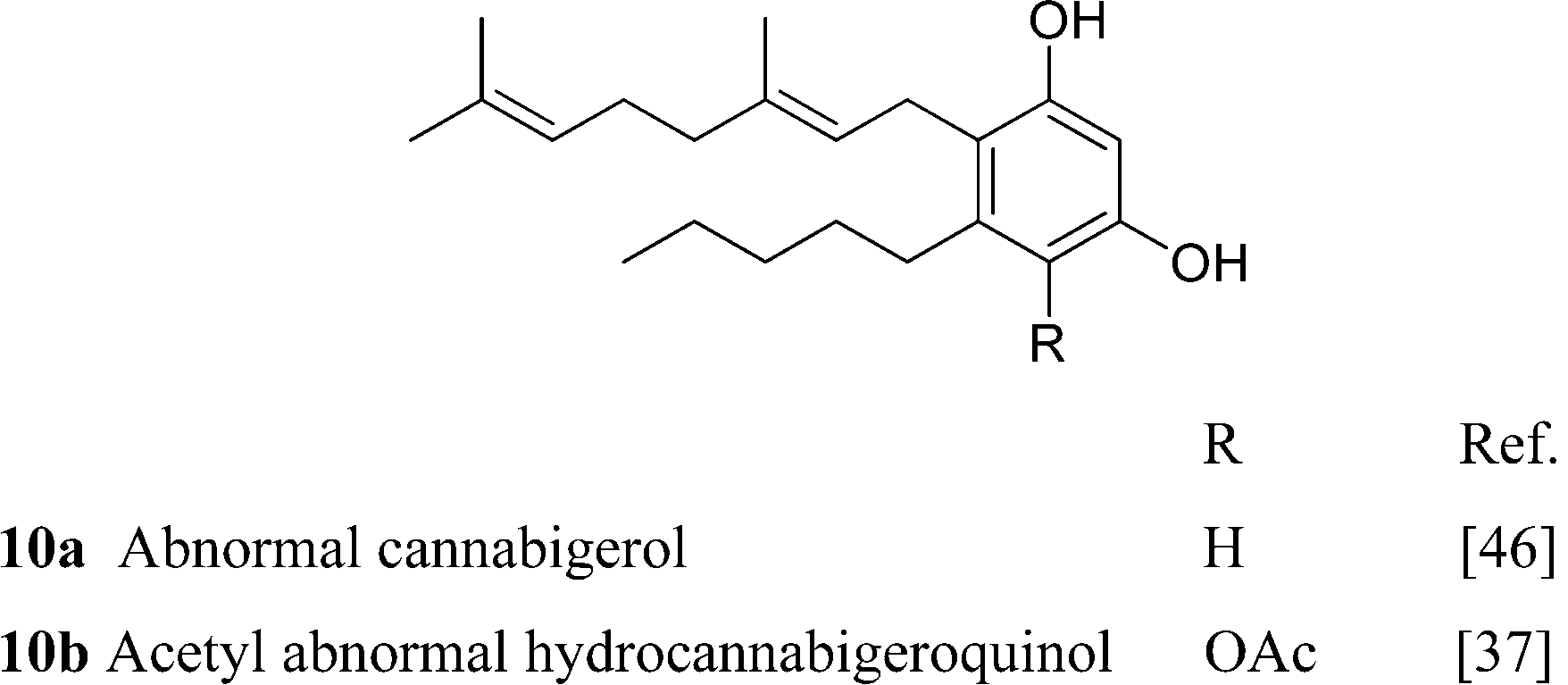
It is not clear if the various oxidized versions of cannabigerol are natural products or rather isolation artifacts. The geranylation of olilvetol gives a mixture of CBG and its positional isomer, the so called “abnormal” cannabigerol (10a).15 While abnormal cannabigerol has never been reported from cannabis and only occurs in H. umbraculigerum, both its actylated hydroquinol (10b) and quinol (11) forms have been detected in a high potency Δ9-THC-strain.37 The only sesquiterpenyl cannabinoid isolated so far belongs to the cannabigerol series, but it is likely that sesqui-cannabinoids also occur in other structural types biogenetically derived from linear isoprenyl cannabinoids.6 The deprenyl derivative of O-methylcannabigerolic acid (amorfrutin 2, 7), a constituent of leguminous plants (see 4.2.1), is one of the few n-pentyl-type phytocannabinoids not isolated from cannabis.54
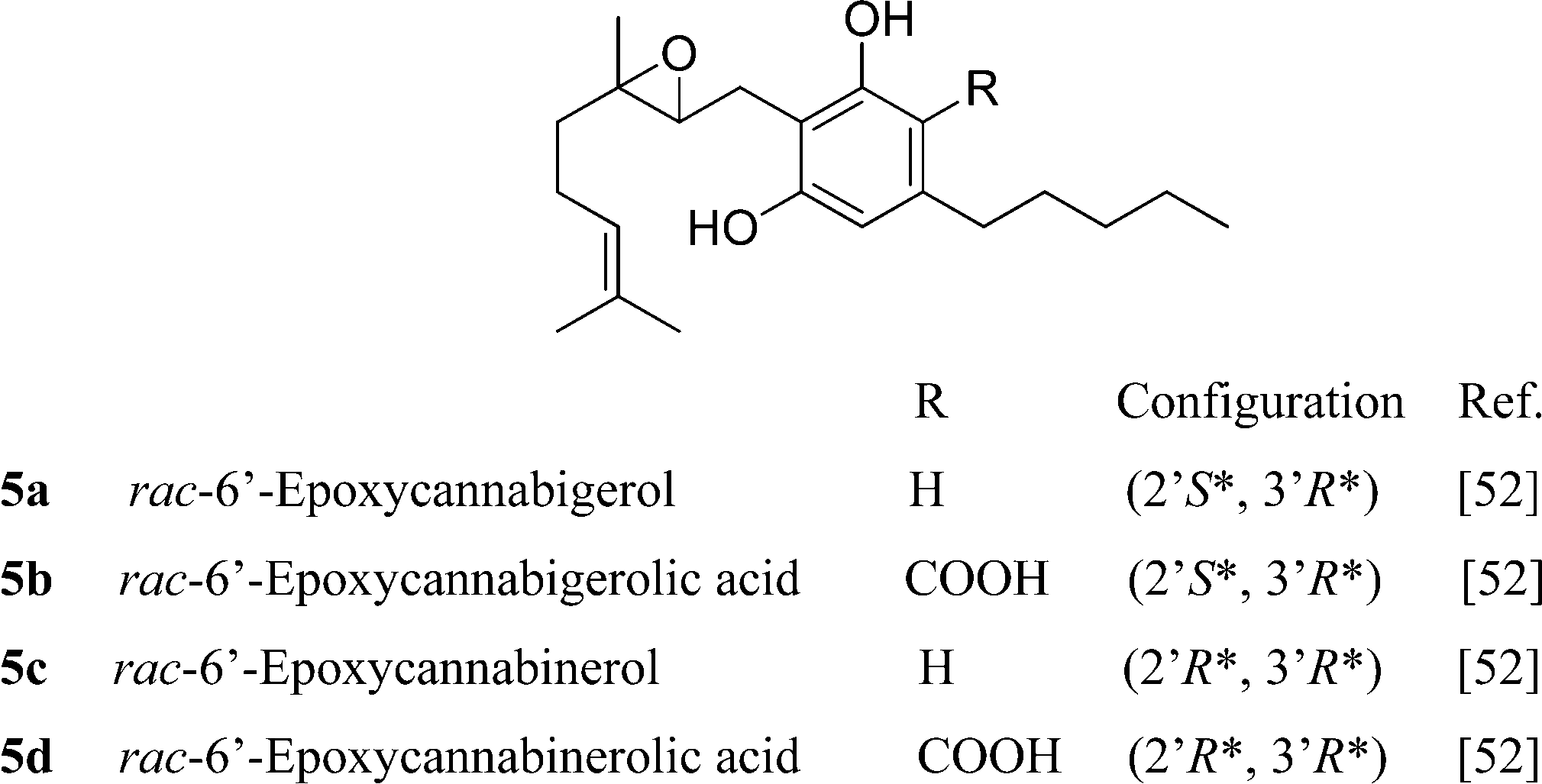
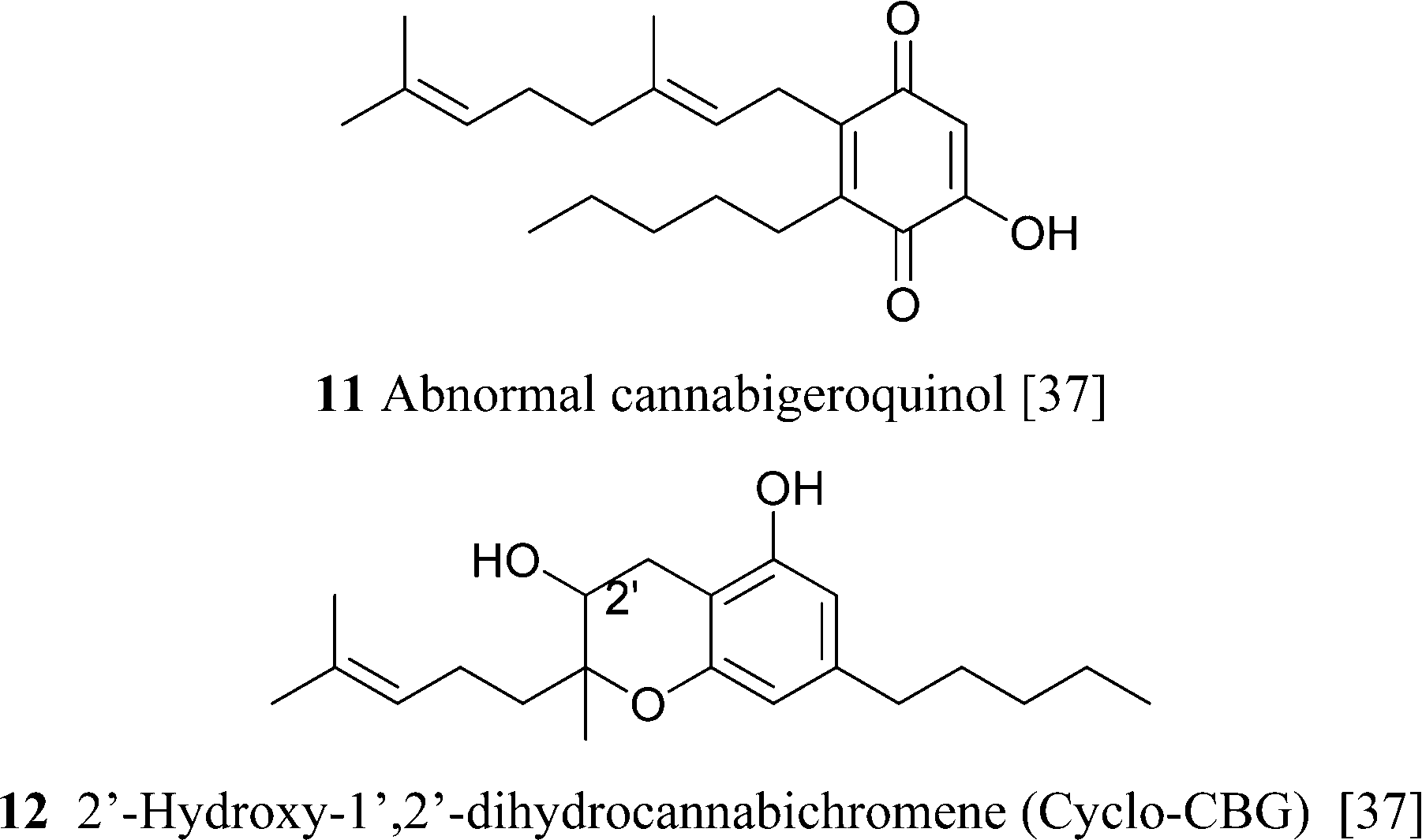
CBG is unstable to acids and bases. Mineral acids cyclize the terpenyl moiety,15 while in strong bases (heating with BuLi in HMPA), the proximal (Δ2′) double bond is isomerized to the phenyl-conjugated E-Δ1′-isomer, a reaction mediated by deprotonation at C-1′.55 Removal of the benzylic proton might involve proton transfer mediated by a phenate ion, since bis-O-methyl CBG was stable in these conditions (Scheme 4).55 Compound 12,37 although structurally a chromene, is most likely derived from the intramolecular cyclization of ω-epoxycannabigerol, a compound so far unknown from natural sources, and, as a cyclo-CBG, is therefore included in this group of phytocannabinoids.
 |
||
 |
||
 |
||
 |
||
 |
||
 |
||
 |
||
 |
||
 |
||
 |
||
| Scheme 4 Isomerization of CBG in basic conditions. | ||
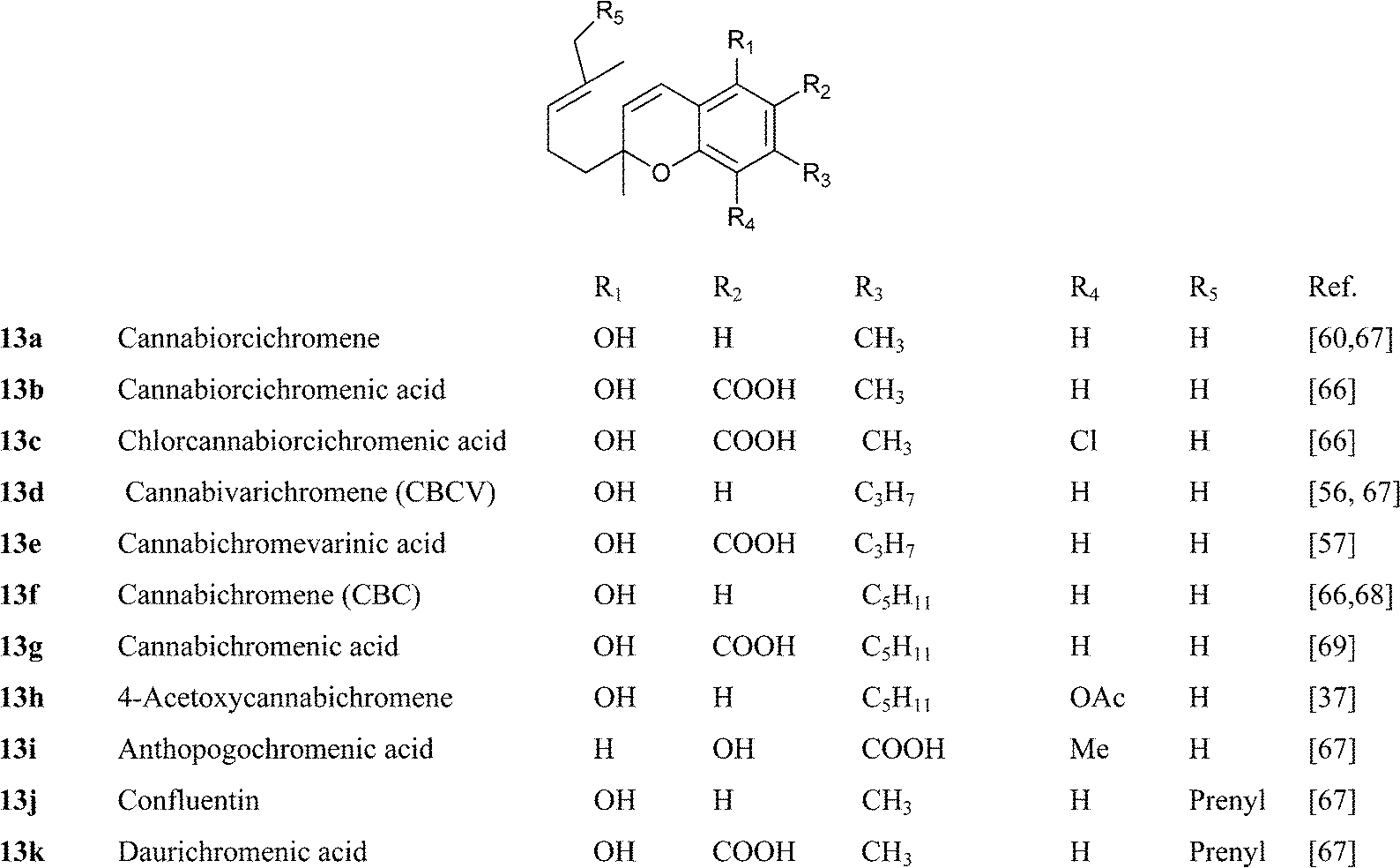
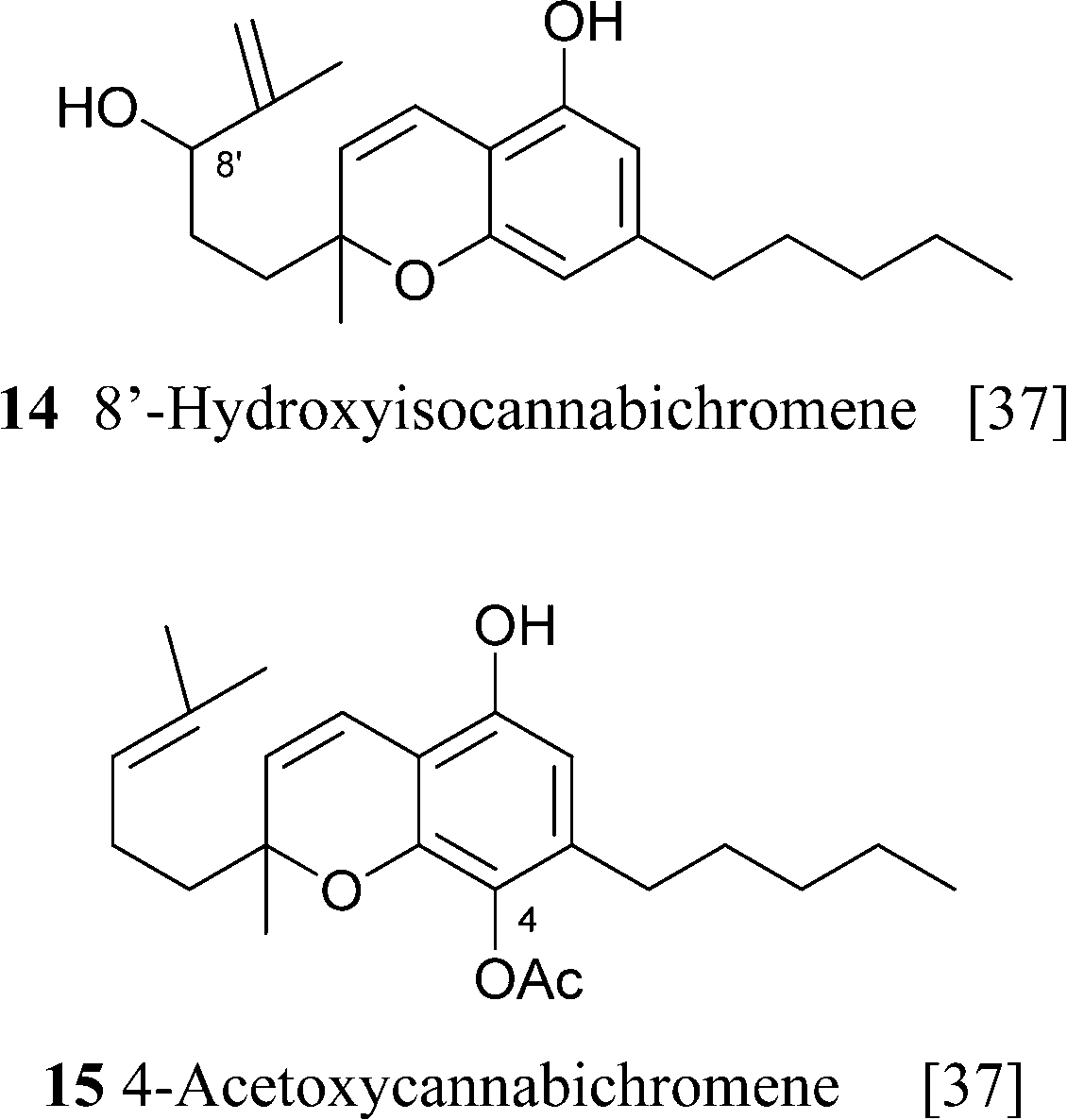
CBC is the simplest natural phytocannabinoid to obtain by synthesis, being available, apart from CBG by oxidative dehydrogenation, also from the one step condensation of citral and olivetol (see Section 4.1.5 for a discussion on the mechanism of the reaction).65 CBC is stable, and has been detected in century-old historical samples of cannabis.31 As with CBG, diversity in the derivatives of CBC is associated to oxidation of the prenyl group and the aromatic ring, with the hydroquinol hydroxylation pattern being stabilized by acetylation. The configurational aspects of hydroxylated cannabichromenes 14 and 16 have not been elucidated. Since natural CBC is racemic, these compounds are most probably a mixture of diastereomers. Remarkably, the orcinol-type cannabichromenes 13b and 13c are of fungal and not plant origin, and have been obtained from Cylindrocarpon olidumWollenw., a parasite of the root knot nematode Meloidogyne incognita, a major pest of some cultivated plants,66 while the sesquicannabinoids confluentin (13k) and the anti-HIV agent daurichromenic acid (13l) have been isolated from a rhododendron species (Rhododendron dauricum L.) with confluentin having also been reported as a constituent of the mushroom from the genus Albatrellus.67 In accordance with the racemic nature of CBC, confluentin (13j) was reported as a racemate, while daurichromenic acid (13k) as well as several functionalized analogues were isolated in an optically active form.67 This suggests that racemization via an electrocyclic mechanism might be slowed by the presence of a carboxylic group para to the chromenic oxygen.
 |
||
 |
||
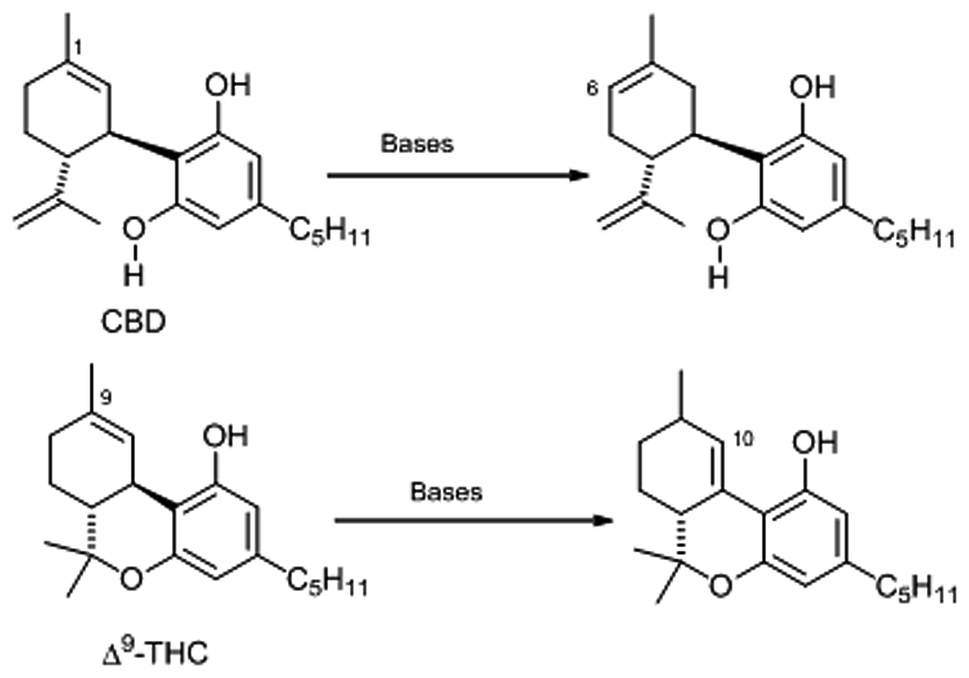
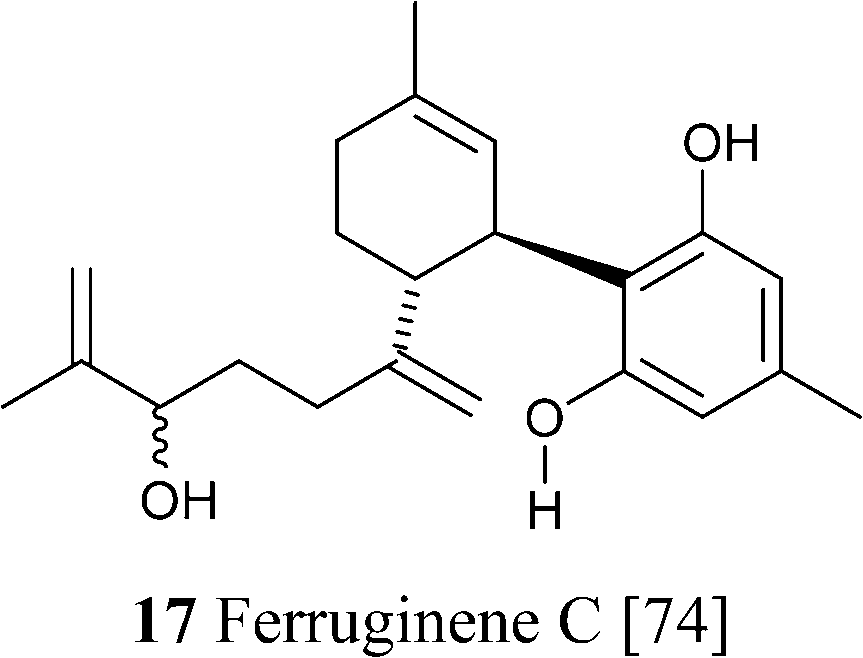
The isolation of a prenylogue orcinoid analogue of CBD (17) was reported from the Alpine rhododendron (Rododendron ferrugineum L.).74 This compound showed only negligible affinity for CB1 and CB2, not unlike CBD.74 Despite the structural similarity between CBD and Δ9-THC, the two compounds show a distinct biological profile, and, even though CBD can be electrophilically cyclized to Δ9-THC by treatment with acids,65 the two compounds are the result of independent oxidative cyclizations of their common precursor CBGA, and are not interconverted in cannabis tissues.29 Δ9-THC and CBD have also quite different oxidative stability. While THC is roughly planar and removal of the benzallylic proton (H-10a) leads to a conjugated radical, the two rings of CBD lie in different planes,75 and the benzyl radical generated from CBD cannot therefore benefit from conjugation with the aromatic ring. The slow (relatively to the NMR time scale) rotation around the terpenyl–resorcinyl bond is an interesting case of aryl-C(sp3) hindered rotation en route to atropisomerism, and is responsible for the temperature-dependence of the NMR spectra of CBD.75 The impossibility to attain planarity and conjugation due to E-strain is also responsible the different behaviour of CBD and Δ9-THC in bases. While the latter generates the conjugated Δ10 isomer, CBD is isomerized to its further de-conjugated Δ6-isomer, a compound of unknown bioactivity (Scheme 5).55
 |
||
| Scheme 5 Base-catalyzed isomerization of CBD and Δ9-THC. | ||
The acid-catalyzed cyclization of CBD to a mixture of narcotic THC isomers might be of relevance for the biological profile of CBD, rationalizing, for instance, the high incidence of somnolence observed in pediatric studies.76 In simulated gastric fluid (pH = 1), the conversion of CBD, solubilized with sodium dodecyl sulfate, to a mixture of Δ9 and Δ8-THC was 98% complete after 2 hours, although the insolubility of CBD might slow down the reaction under physiological conditions.76 This could also rationalize the observation that CBD is unable to generate significant amounts of Δ9-THC on smoking marijuana,77 whose water suspensions are mildly basic (pH ca. 8). On the other hand, CBD can do so in the more acidic (pH ca. 5.7) tobacco cigarettes when they are spiked with CBD or CBD-containing cannabis oil, a popular practice within cannabis consumers.3 The pyrolysis of CBD under conditions mimicking smoking gave a complex mixture of products. Apart from small amounts of Δ8– and Δ9-THC, the major products identified were cannabielsoin (39c) and its C-1 epimer.78
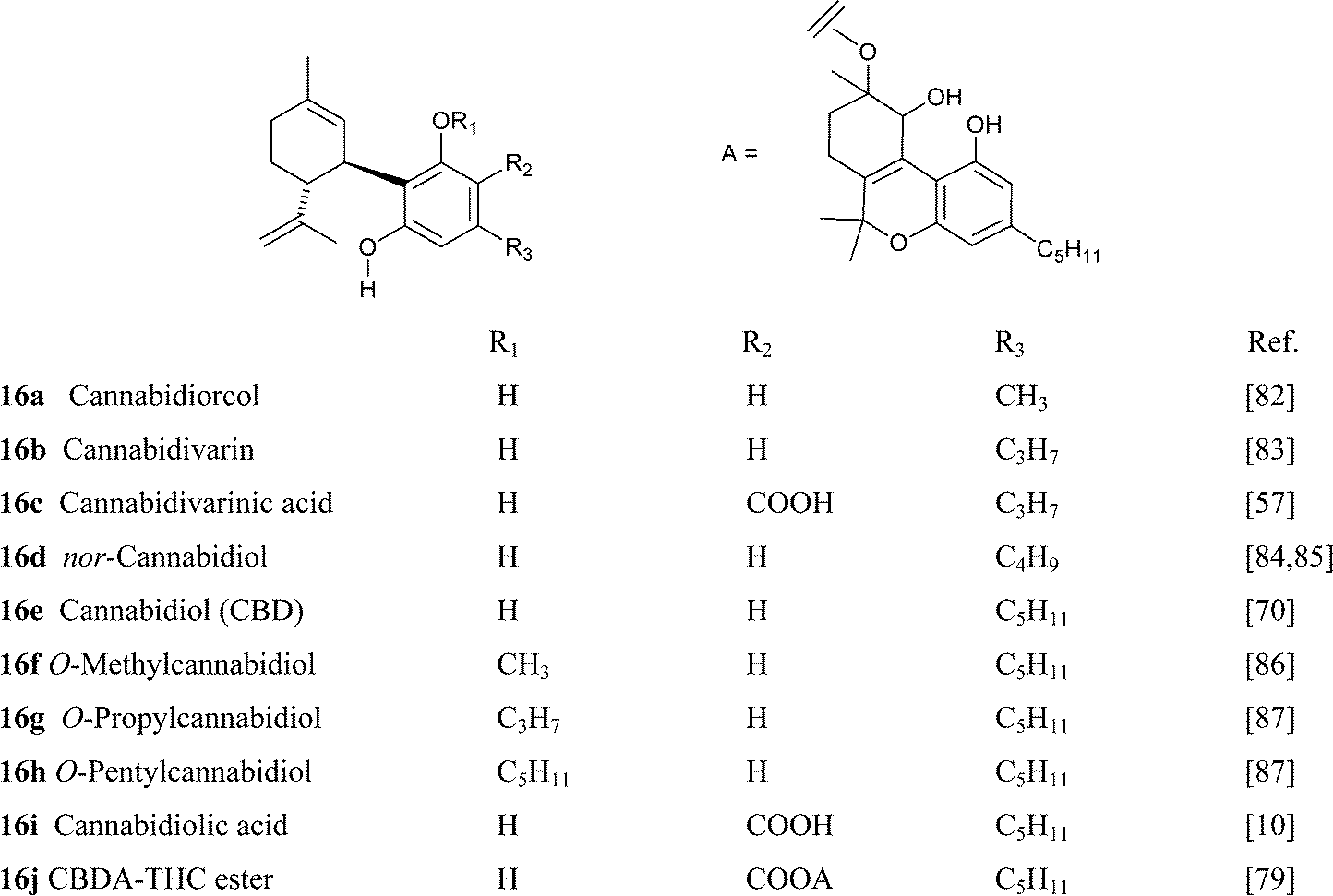
Some of the naturally occurring analogues of CBD show interesting structural features, like the presence of an alkyl residue with an even number of carbons (nor-CBD, 16d) or O-alkylation with propyl- and pentyl residues. The isolation of an ester of cannabidiolic acid with a dihydroxylated Δ6a,10a-tetrahydrocannabinol derivative (16j) has also been reported. This compound was the first complex ester of pre-cannabinoids to be isolated.79
CBD is an allosteric inhibitor of CB1,80 and further modulates the activity of Δ9-THC by interfering with its hepatic allylic hydroxylation, a reaction that generates a metabolite (11-hydroxy Δ9-THC) with a higher brain penetration and similar potency on CB1.39 Despite the enormous current interest for the clinical uses of CBD, the first studies for the bioactivity of CBD were actually triggered by its modulating activity on cytochromes and the potential for drug interaction, with the synergizing activity of CBD on the hypnotic effects of barbiturates being already reported in 1942 by Adams himself.39 CBD seems to have a host of biological targets, including various thermos-TRP channels and the serotonin receptor 5-HT1A,64 and its overall biological profile cannot probably be summarized by the modulation of any single end point of the growing list of CBD biological targets. Currently, the major area of clinical research on CBD is the management of pediatric epilepsy, a use reminiscent of the first report on the medicinal use of Cannabis in colonial India by W. B. O’Shaughnessy in 1838.81
 |
||
 |
||
 |
||
 |
||
 |
||
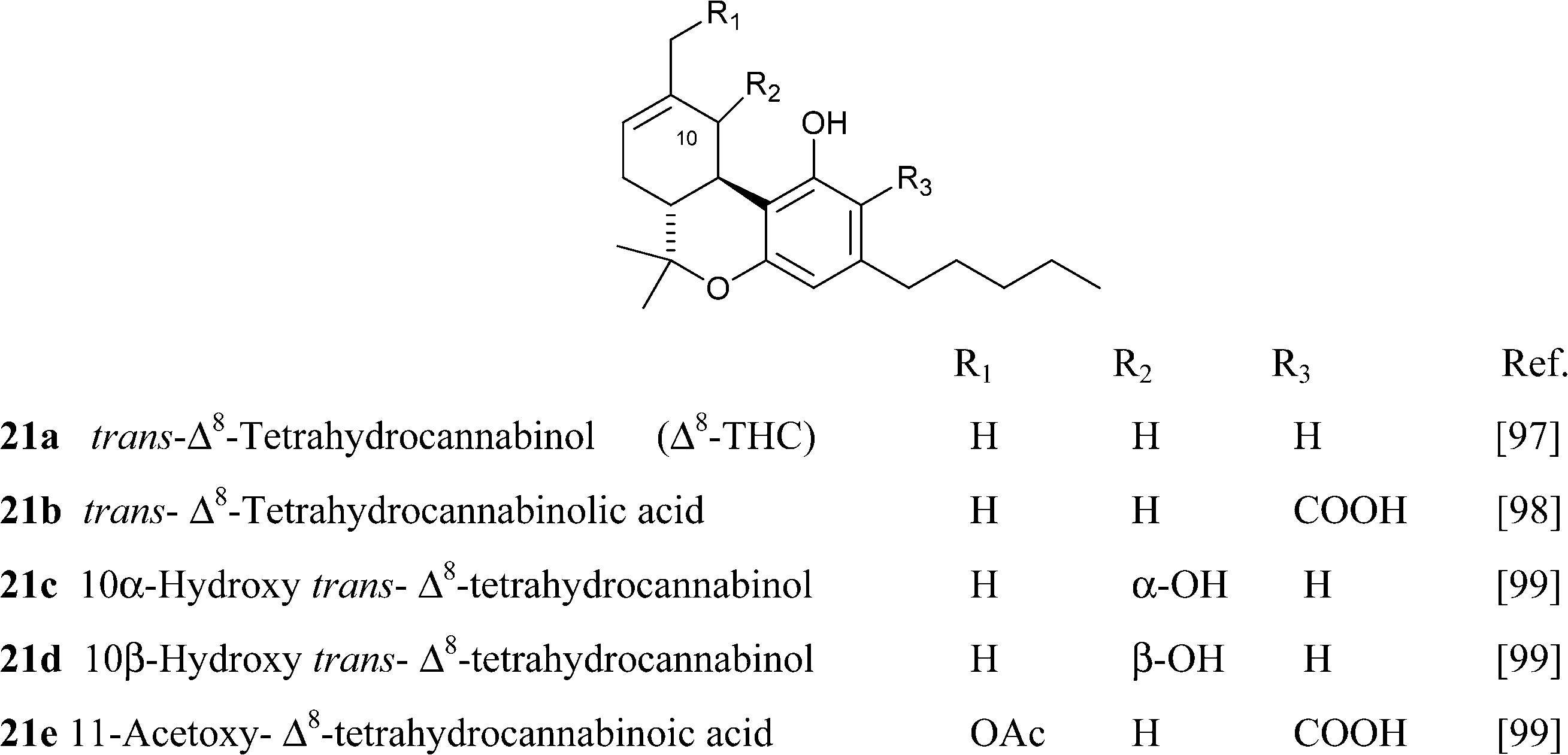
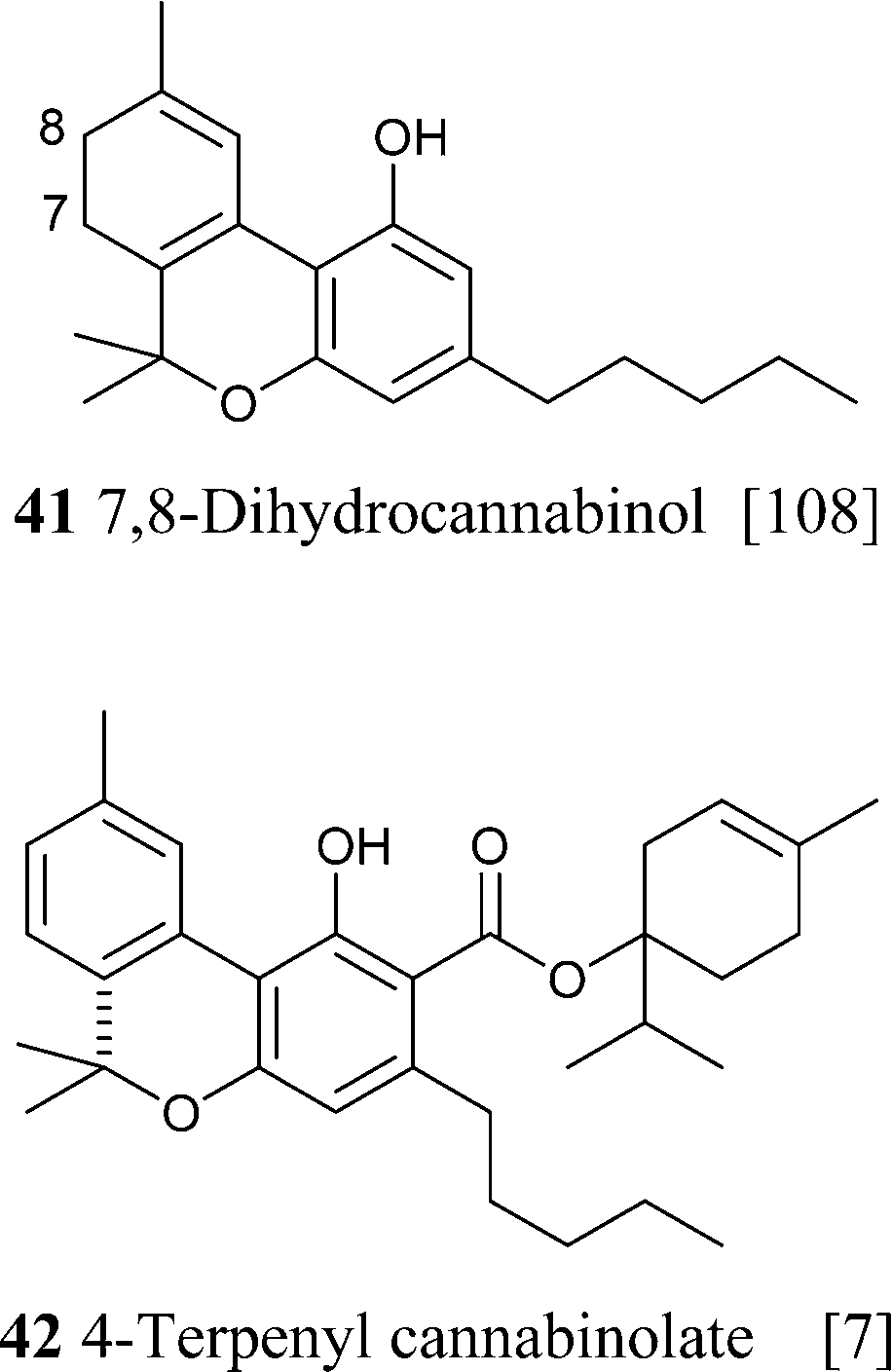
4.1.5.1. Δ8-tetrahydrocannabinol (Δ8-THC)-type compounds. Compounds of this class might be isolation artifacts resulting from Δ9-THC by acid- or oxidatively promoted shift of the endocyclic double bond, or from CBD by electrophilic cyclization. The Δ8 location is thermodynamically more stable than the Δ9 location, and this drives the isomerization.65 The major spectroscopic difference between the two isomeric series is the chemical shift of the olefinic proton, that, because of the proximity to the aromatic ring, is more deshielded in the Δ9-isomer (δ ca. 6.40 in CDCl3) compared to the Δ8-isomer (δ ca. 5.50 in CDCl3).93 The electrophilic cyclization of CBD can afford the Δ8– or the Δ9-isomer depending on the conditions, with mild acidic conditions favoring the Δ9-isomer and more forced conditions in terms of acidity and temperature the Δ8-isomer.93 Δ8-THC and Δ9-THC show a similar profile of activity on cannabinoid receptors, with Δ8-THC being only slightly less active than Δ9-THC.45 It should, however, be interesting to evaluate the profile of the two isomers also in terms of other targets, like thermo-TRPs and transcription factors of the PPAR family, since this could provide interesting clues to clarify the role of the non-metabotropic targets in the pharmacological profile of Δ9-THC.
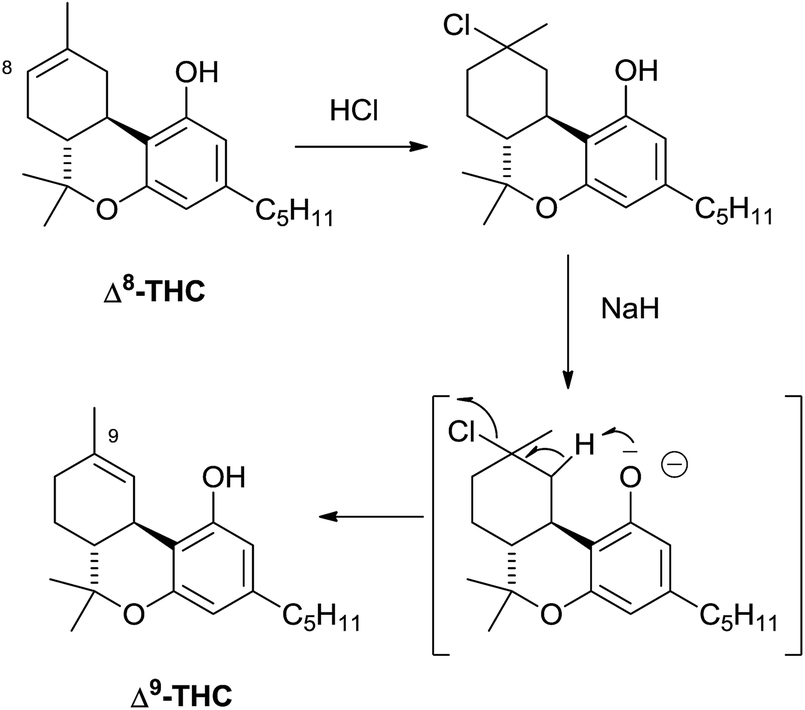
Compounds from the Δ8 series can be converted into their Δ9 isomers by addition of hydrochloric acid and base-mediated dehydrohalogenation (Scheme 6).94 The counter-thermodynamic course of the reaction has been rationalized by assuming that deprotonation occurs intramolecularly via a phenate ion, thus favoring deprotonation from C-10 rather than from the other carbons adjacent to C-9.94 This reaction is of great relevance, since Δ8-THC is much easier to synthesize than Δ9-THC (one step from verbenyl olivetol).95 The isolation of a compound oxygenated at C-11 is interesting, since this is a major route in the human metabolism of Δ9-THC. In general, compounds from the Δ8-series are much more stable than their Δ9-series, and Δ8-THC has even been detected in a burial tomb dating from the fourth century B.C.96 Because of the improved stability compared to Δ9-THC and its easier synthesis, Δ8-THC proved a better lead structure for phytocannabinoid-inspired probes to explore the biological space around cannabinoid receptors.45
 |
||
 |
||
 |
||
| Scheme 6 Conversion of Δ8-THC into Δ9-THC. | ||
4.1.5.2. Δ9-trans-tetrahydrocannabinol (Δ9-THC)-type compounds. The early investigations on the phytochemistry of cannabis came to the conclusion that the narcotic constituent of the plant was a reduced form of cannabinol, at that time the only cannabinoid whose structure was known. The nature of this “active” tetrahydrocannabinol, possibly confusingly purified as acetyl derivative already in 1942,101 remained elusive and confusing until the seminal paper by Gaoni and Mechoulam who in 1964 disclosed its isolation and structure elucidation from a Lebanese sample of hashish.102 Curiously, the Δ9-location of the double bond turned out to be the only one never considered in all the previous investigations on the elusive narcotic principle of cannabis.102 As with CBD, Šantavý came independently to the same conclusions, also establishing the absolute configuration of the active narcotic principle by correlation of Δ9– and Δ8-THC with CBD.72 Δ9-THC belongs to the largest class of phytocannainoids, but the investigation on the phytochemistry of cannabis was long biased on the recreational chemotypes, and future studies on fiber hemp might reveal a different scenario. Diversity within this class of phytocannabinoids is mostly related to oxidation of the p-menthene moiety, possibly related to spontaneous degradation of the natural product (see infra), and to the esterification of pre-THC with various isoprenyl alcohols.
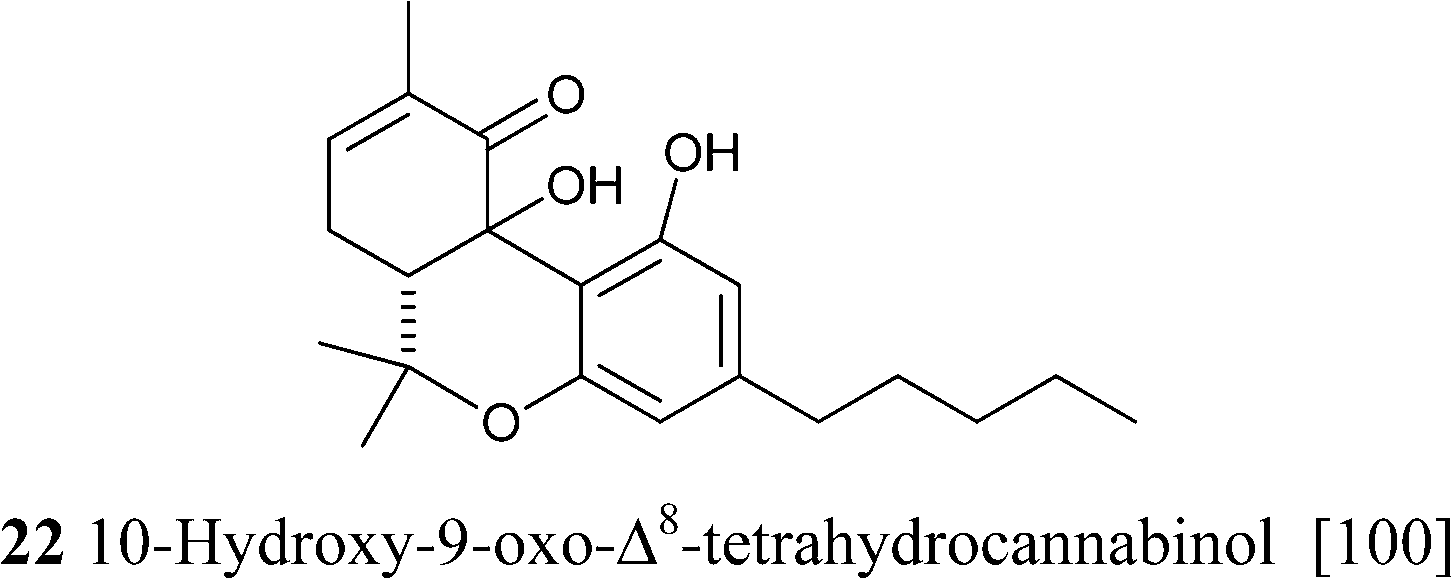
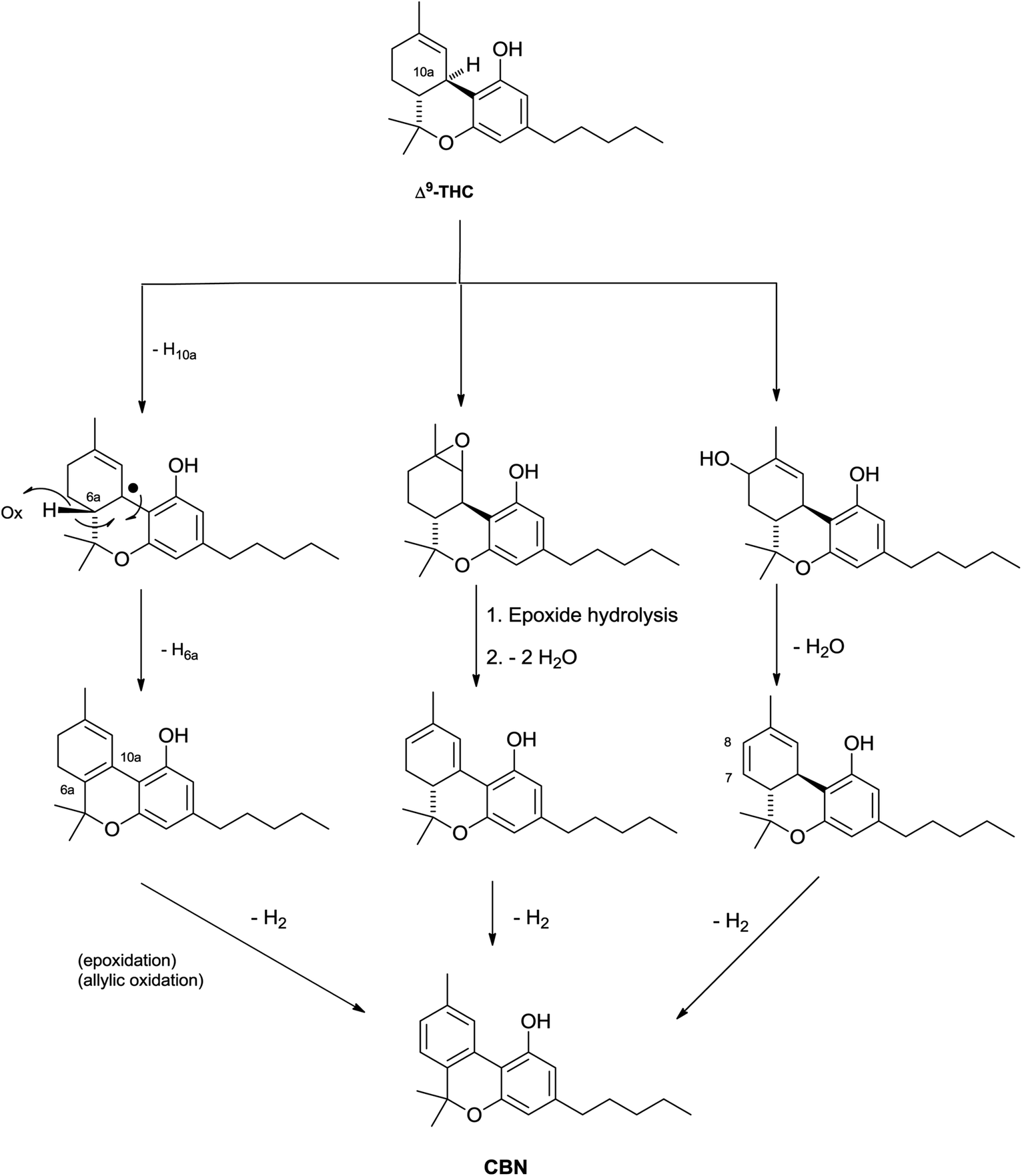
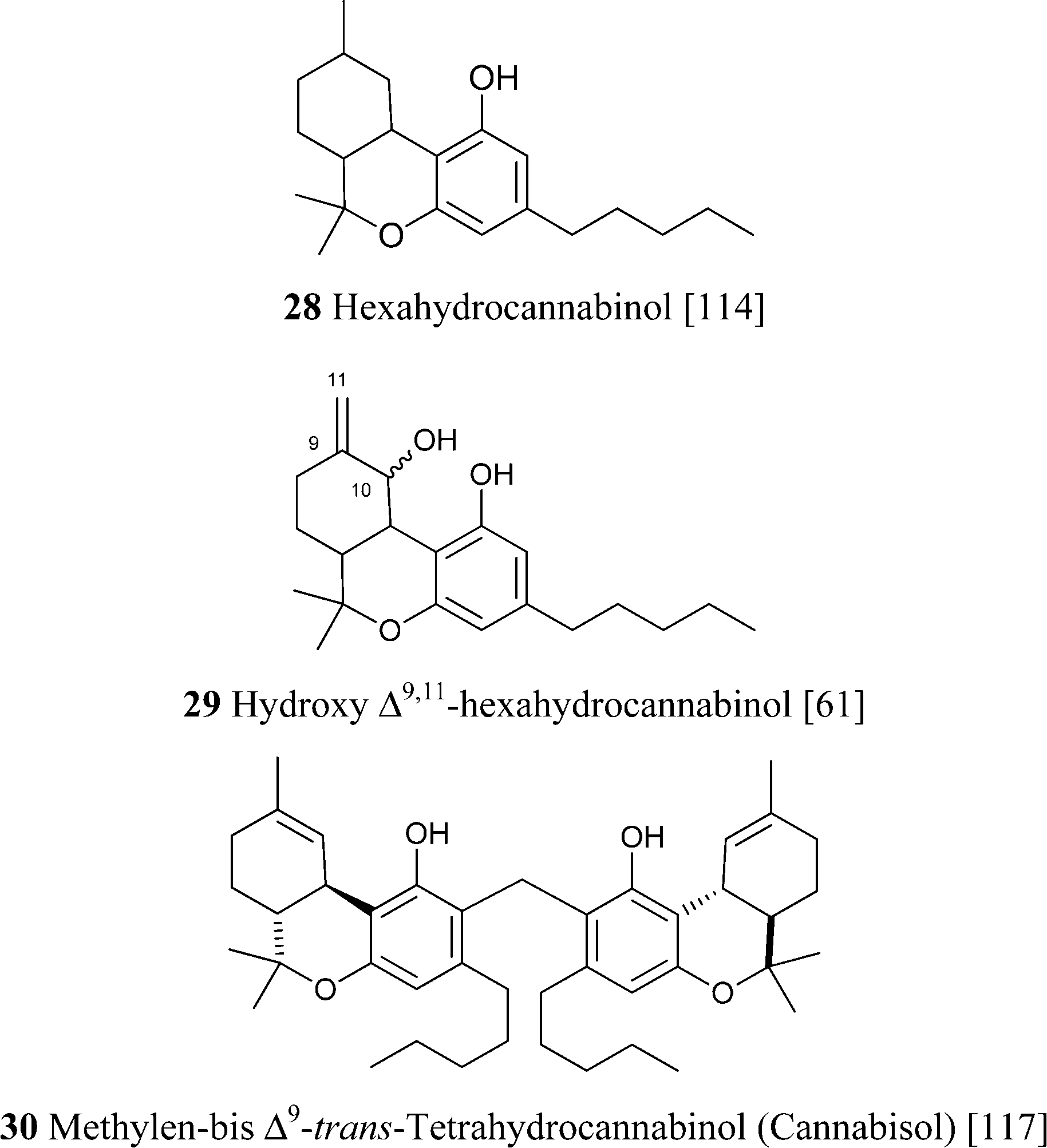
Δ9-THC acts as a partial agonist at both CB1 and CB2,45 but, unexpectedly, its shorter analogue from the bis-nor type (THCV, 23c) is instead an antagonist at CB1, an important discovery in the light of the observation that rimonabant and most synthetic inhibitors of CB1 are actually reverse-agonist and not antagonists.103 The phenolic hydroxyl is critical for the activity, but, surprisingly, branching in the alkyl residue makes it redundant for the interaction with CB1.104 The native form of Δ9-THC is represented by a mixture of two pre-cannabinoids, THCA-A and THCA-B, very different in terms of physical state (THCA-B was investigated by crystallographic studies, while THCA-A is amorphous), stability toward decarboxylation (THCA-A is decarboxylated at 90 °C, while THCA-B is stable at this temperature), and concentration in plant tissues.105 The acidic form of Δ9-THC-A is stabilized toward decarboxylation by esterification with isoprenyl alcohols, and these conjugates occur, as a complex mixture, in narcotic cannabis.37 The structure of these compounds was only tentatively assessed, and the configuration of the isoprenyl residue should be confirmed by an independent synthesis. Δ9-THC is unstable as a pure compound, an amorphous gum that easily turns brown, but is more stable in crude form and can be stored in refrigerated methanol solution. The degradation is mainly oxidative, and is triggered by abstraction of the allylic and benzylic hydrogen at C-10a (Scheme 7). The resulting radical undergoes further hydrogen abstraction at C-6a, with formation of a conjugated double bond between C-6a and C-10a, en route to aromatization to CBN. Alternative dienes can be generated via either epoxidation of the endocyclic double bond, hydrolysis of the epoxide, and twofold dehydration, or via allylic oxidation at the C-8 methylene and dehydration. Aromatization of these dienes eventually generates CBN (Scheme 7).106 At room temperature, the rate of degradation of Δ9-THC in cannabis has been estimated in ca. 5% per month, and 10% for the pure product, but other degradations pathways have been postulated be operative in plant tissues, since the rate of appearance of CBN was significantly lower than the one of disappearance of Δ9-THC.106 On the other hand, this discrepancy could be related to the quick formation of intermediates that then converge to CBD at a slower rate. The mechanistic scenario for the aromatization is in accordance with the isolation of some of the intermediate compounds as well as with the detection of radicals by electron spin resonance during the degradation process.107 There are no recent studies on the degradation of Δ9-THC, and the development in analytical technology witnessed by the past decades should greatly help the clarification of this important process. Interestingly, a tri-hydrocannabinol (28) has been isolated from the pollen of cannabis.108 This compound could originate by disproportion of a dihydrocannabinol intermediate.
 |
||
| Scheme 7 Possible mechanism for the oxidative degradation of Δ9-THC to CBN. | ||
The acidic isomerization of Δ9-THC generates the thermodynamically more stable Δ8-isomer, that does not undergo oxidative degradation either in plant material or as a pure product, in accordance with the minor stabilization by resonance of a C-10a radical, that would now only be benzylic and not benzallylic.109 Δ9-THC is characterized by an extremely low acute toxicity (LD50 > 100 mg kg−1 iv in rats), while CBD and other cannabinoids have a measurable toxicity (LD50ca. 50 mg kg−1 iv in rats for CBD).104
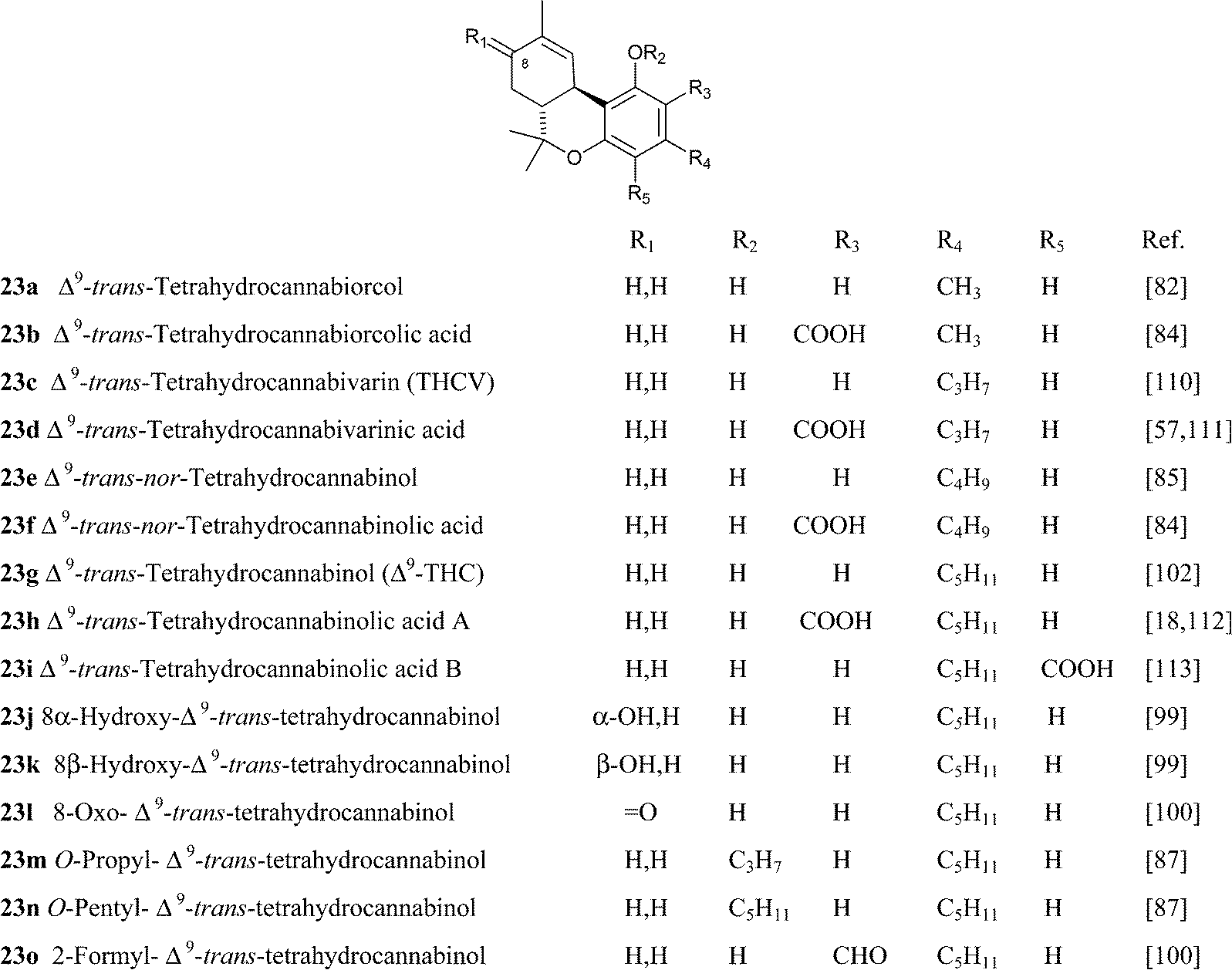 |
||
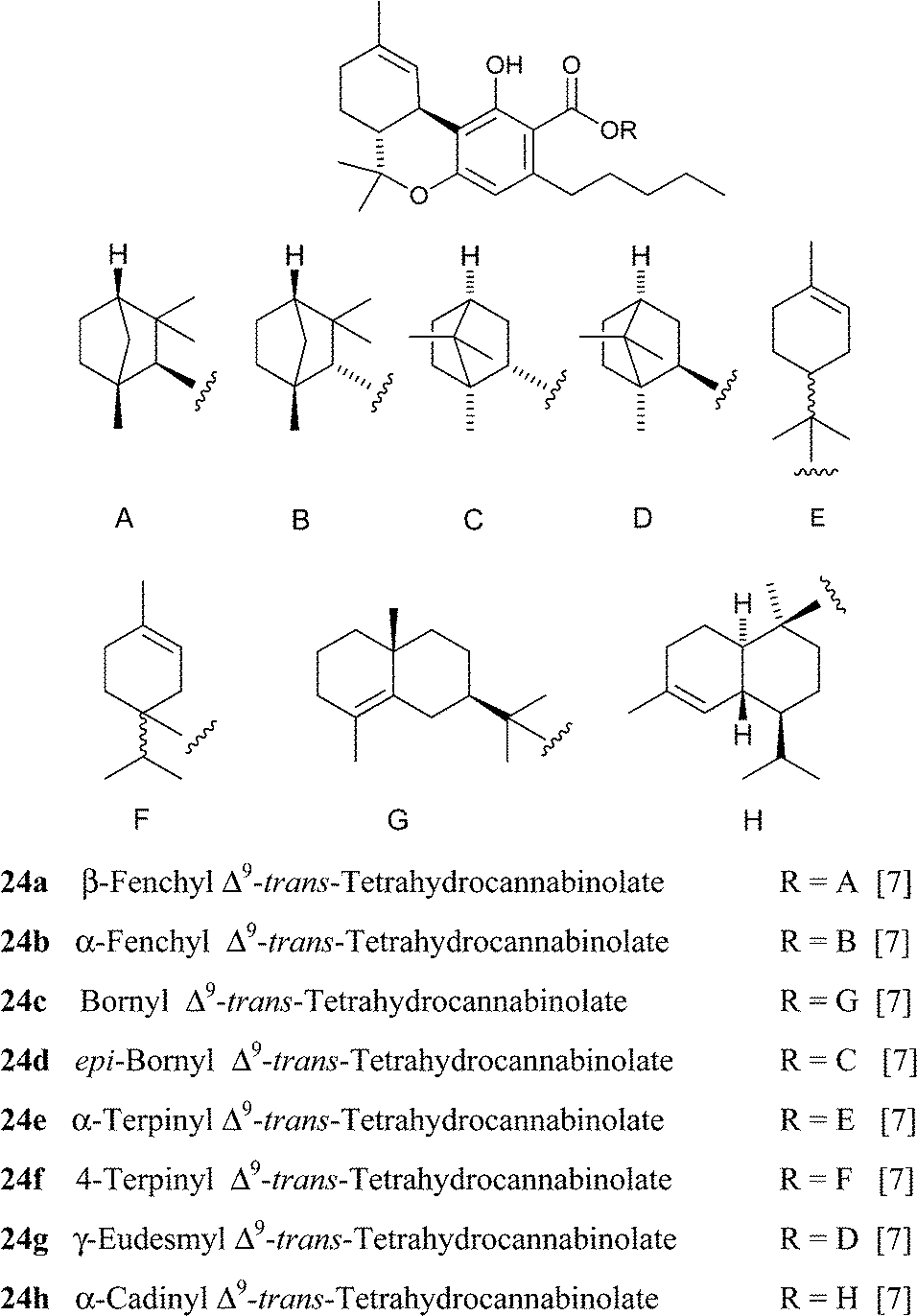 |
||
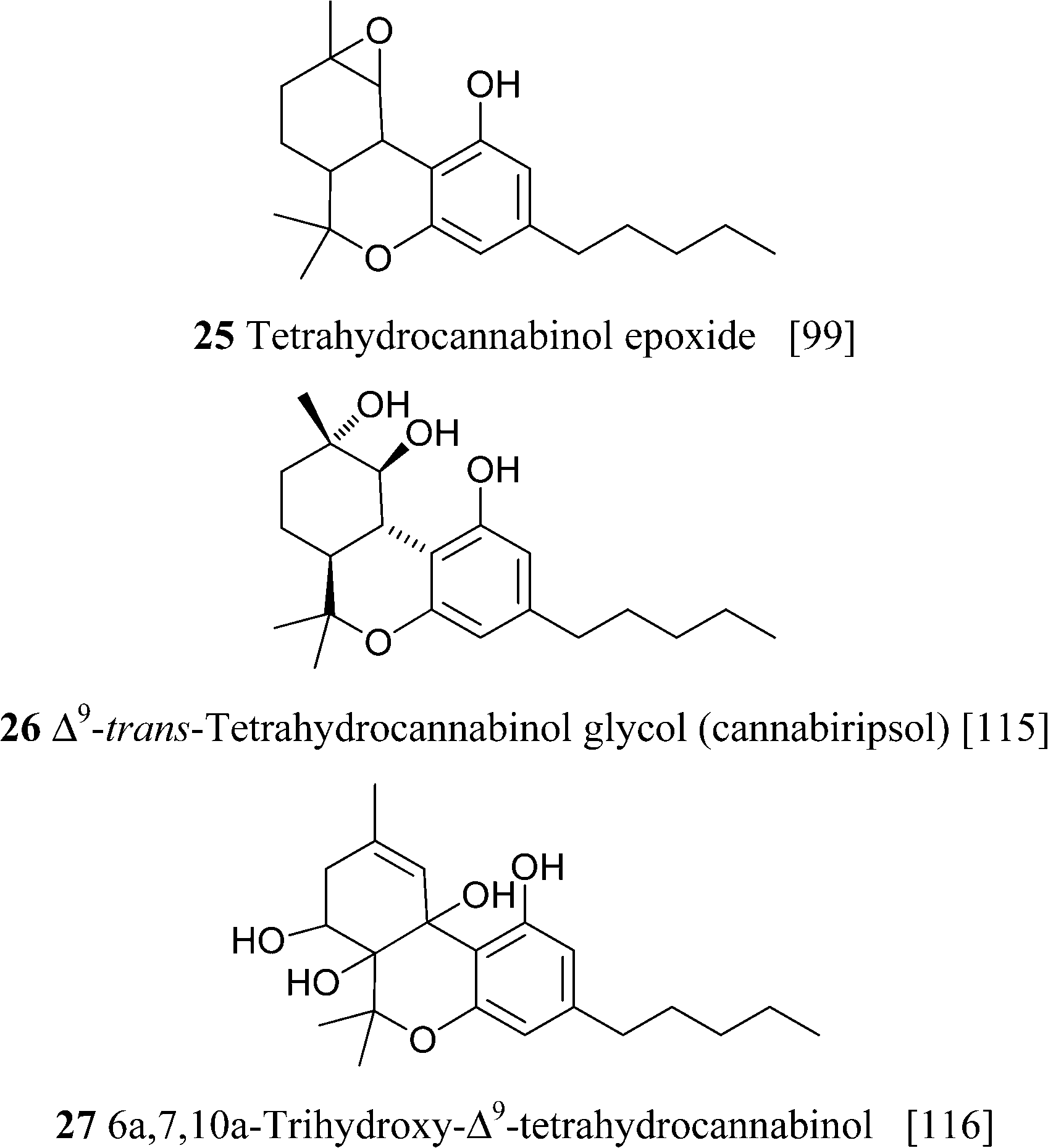
Hydroxylated derivatives of Δ9-THC have been isolated as a diastereomeric mixture, as expected from a non-enzymatic oxidative process. In some cases, as in 27, the configuration at the hydroxylated carbons was not assessed, and it is unclear if the isolated compound was a mixture of isomers or, alternatively, configurationally pure.114 The hydroxylated derivatives of Δ9-THC have been poorly investigated in terms of bioactivity. Interestingly, microsomal hydroxylation of Δ9-THC takes place at the allylic methyl (C-11) rather than at the endocyclic allylic methylene (C-8).45 11-Hydroxy Δ9-THC, unknown as a natural product, substantially retains the affinity of the natural product toward CB1and CB2, but penetrates more easily the brain.45 Also the epoxide of Δ9-THC (25) has been isolated from cannabis,99 while the generation of the methylene-linked dimer cannabisol (30) might be the result of a process similar to the one that forms dicoumarol from 4-hydroxycoumarin.
 |
||
 |
||
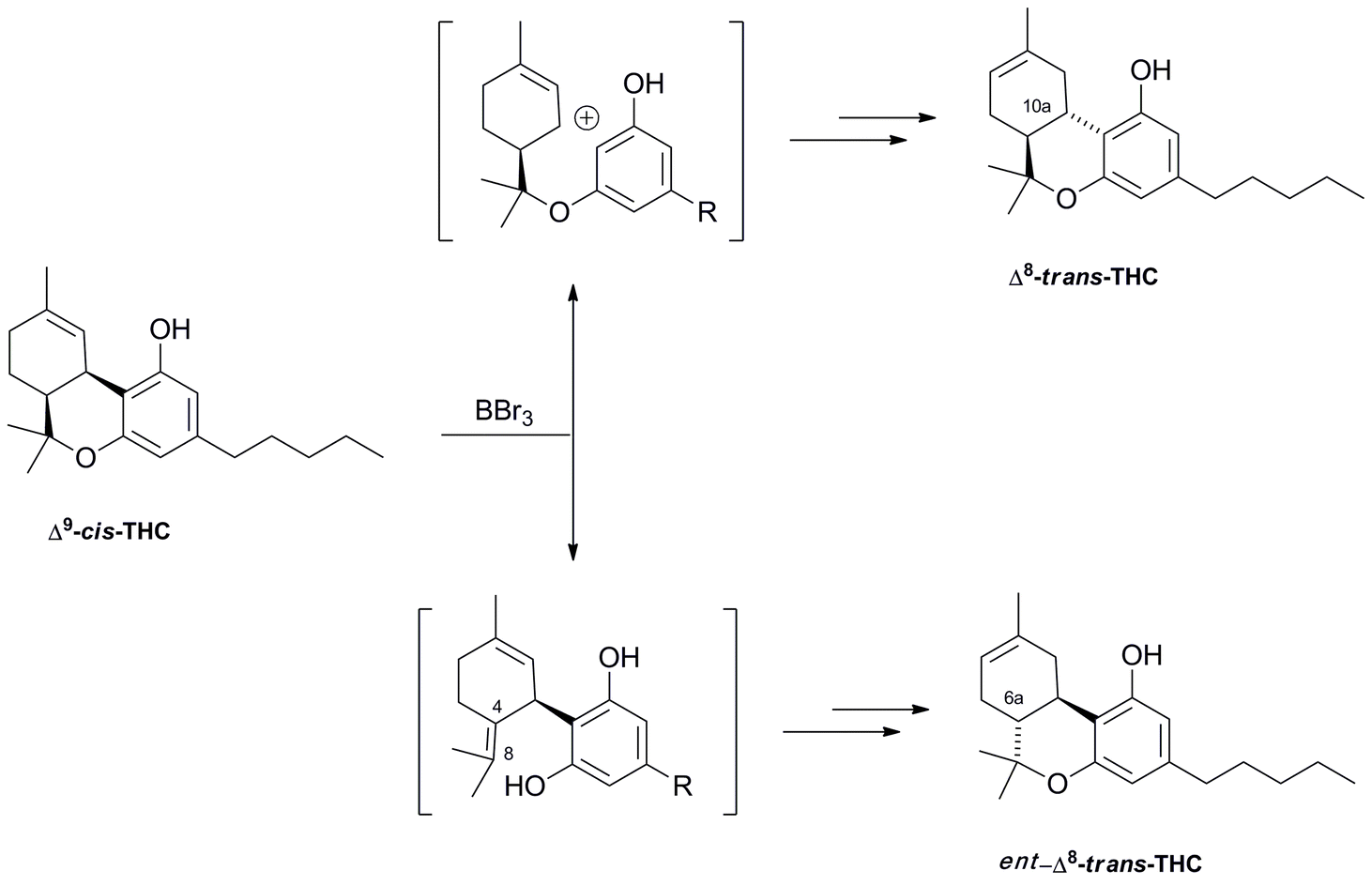
4.1.5.3. Δ9–cis-Tetrahydrocannabinol-type compounds. The existence of a cis-isomer of Δ9-THC in cannabis has long been known, but the structure of this compound is still unclear, and the confusing history of this compound exemplify the subtleties of cannabinoid chemistry. Δ9–cis-THC is only a trace constituent of narcotic cannabis, but has been reported to occur in fiber hemp in concentrations similar to those of its trans-isomer, an important observation waiting, however, confirmation in modern studies.118 Since the presence of significant amounts of Δ9–cis-THC is associated to the one of large amounts of CBD, it is not unconceivable that Δ9–cis-THC could actually be an artifact, derived by migration of the exocyclic Δ8(9) double bond of CBD to a Δ4(8) position, followed by closure of the pyran ring (Scheme 8). If so, epimerization should be at C-6a (THC numbering), but this reaction has not been clearly observed under laboratory conditions. In accordance with this, treatment with Lewis acids converts racemic Δ9-cis-THC into racemic Δ8–trans-THC, presumably by opening of the oxygen bridge to give a Δ4,8-CBD intermediate, that then re-closes to generate the trans-isomer (Scheme 8).119 However, under these conditions, interconversion from the normal- to the abnormal series has also been observed, showing that also the cleavage of the resorcinyl-menthyl bond via a retro-Friedel Craft reaction is, in principle, possible.119 By using optically active substrates, it was eventually demonstrated that the isomerization takes place via cleavage of the pyrane ring, but it is unclear how this relates to the configuration of natural Δ9–cis-THC, if this is, indeed scalemic.119
 |
||
| Scheme 8 Possible mechanisms for the isomerization of cis to transtetrahydrocannabinols. | ||
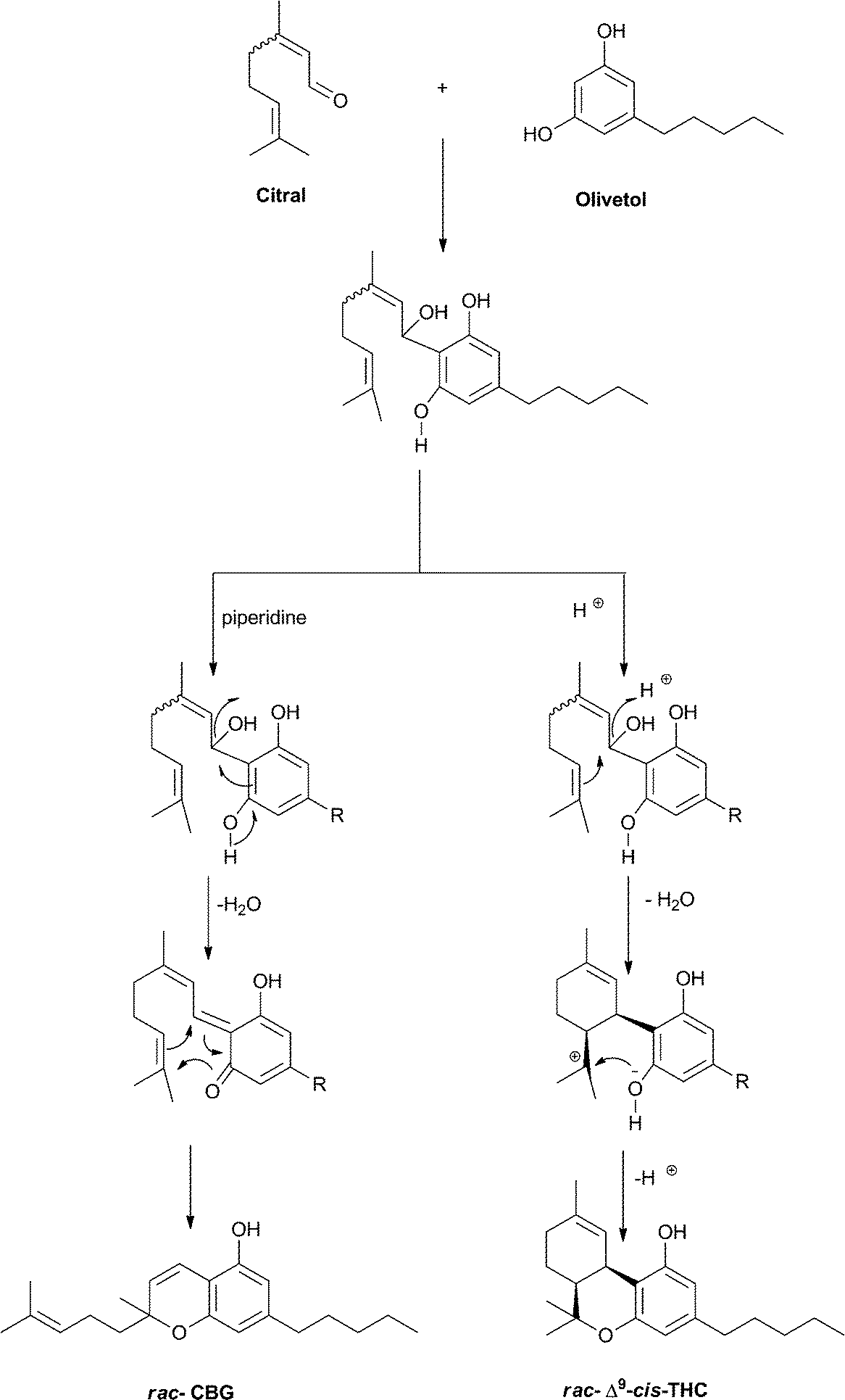
Racemic Δ9–cis-THC can be easily prepared from the condensation of citral and olivetol in acidic medium.93 According to the catalysis, the reaction can afford cannabichromene or Δ9–cis-THC. Presumably, the reaction has a concerted course in basic medium, going through a quinone methide intermediate. Conversely, in the presence of protic or Lewis acids, cyclization of the initial 1,2-adduct to a menthyl cation could occur, followed by cyclization to Δ9–cis-THC (Scheme 9). The relative configuration of the final product depends on the nature of the catalyst. While Broensted acids afford essentially the cis-isomer, Lewis acids selective for the trans-isomer have been developed.120
 |
||
| Scheme 9 Different course of the condensation of citral and olivetol depending on the conditions (R = n-pentyl). | ||
Δ9-Tetrahydrocannabinols from the trans and cis series can be distinguished by the chemical shift of the geminal methyls (Δδ 0.25–0.35 in the trans-series, and 0.08–0.15 in the cis-series) from the signal of the benzylic proton, a broad singlet at around δ 3.50 (CDCl3) for the cis-isomer, and a broad doublet at around δ 3.20 (CDCl3) for the trans-isomer.55,93The profile of bioactivity of Δ9–cis-THC has only been investigated for CB1-related activity, with the epimerization causing a general decrease of activity. The recent development of a stereoselective total synthesis of all isomeric forms of Δ9-tetrahydrocannabinol should make it possible a systematic investigation of the biological translation of the epimerization, as well as a long-awaited evaluation of the configuration of the natural product, if indeed optically active.121
 |
||
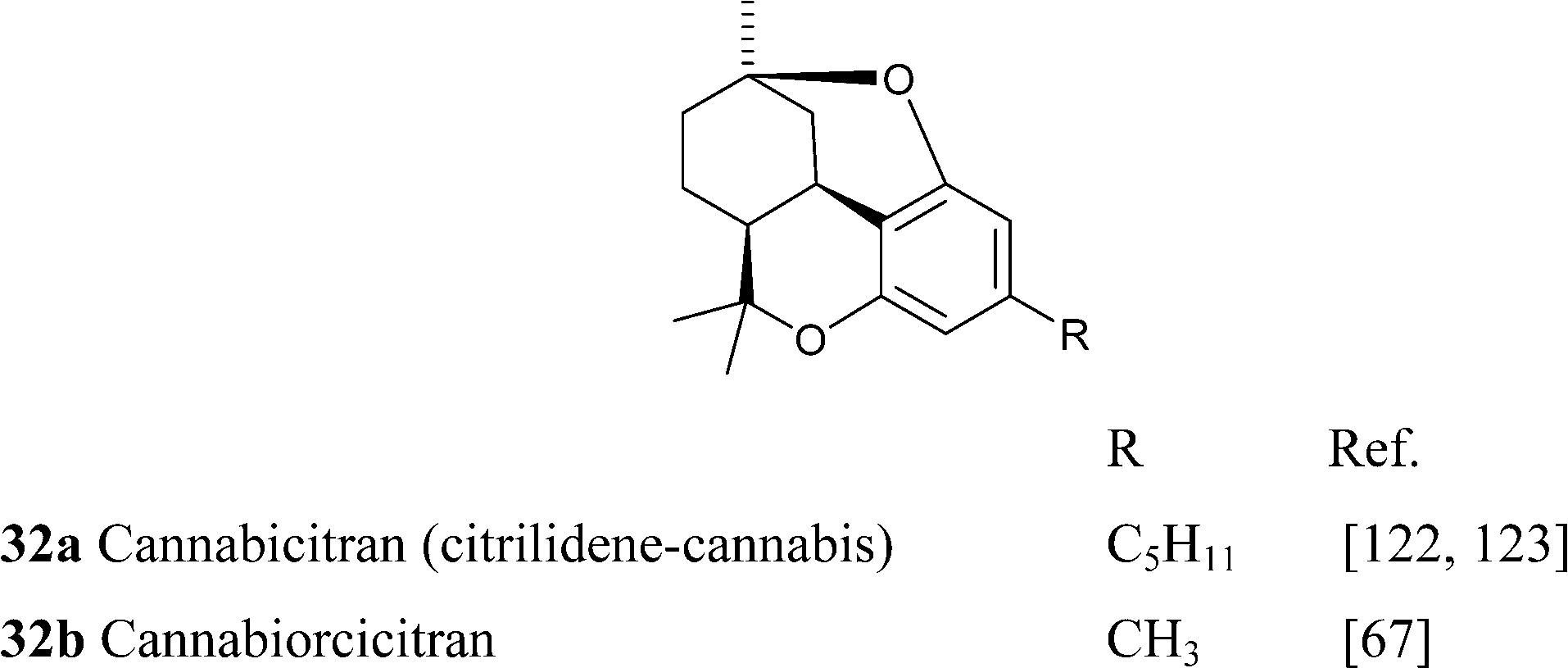
Cannabicitran (32) might derive from cis-THC epoxide by Makovnikov-type protonation of the endocyclic double bond followed by trapping of the tertiary C-9 cation by the free-hydroxyl at C-1. Cannabicitran is an interesting case of “anticipated” natural product, since it was obtained by Crombie122 from the pyridine-promoted condensation of citral and olivetol, before its actual isolation.123 In a rare example of fair play within natural product chemists, Crombie acknowledged the renaming of the compound she had originally named cytrilidene cannabis.
 |
||
4.1.5.4. Δ6a,10a Tetrahydrocannabinol and cannabitriol-type compounds. Compounds of this type are characterized by conjugation between the double bond on the terpenyl moiety and the resorcinyl residue, and are presumably intermediates in the oxidative aromatizatization of Δ9-THC, a process triggered by the generation of a C-10a radical (Scheme 7). Although Δ6a,10a-THC is unknown as natural product, an oxygenated analogue (the epoxide 34) has been isolated from cannabis,108 and the parent compound was synthesized as a racemate by Adams and Todd during the structure elucidation of cannabinol by the preparation of a series of possible putative structures for the natural product.124,125 Racemic Δ6a,10a-THC was found active in the dog ataxia assay, and the observation was confirmed by modern studies, that localized cannabinoid activity exclusively in the S-enantiomer of the racemate.126 The activity was lower, but qualitatively similar to the one of Δ9-THC, and it is therefore surprising that little information exists on compounds of this type, that are stable in ethanol solution and have been detected in historical samples of cannabis tinctures.84
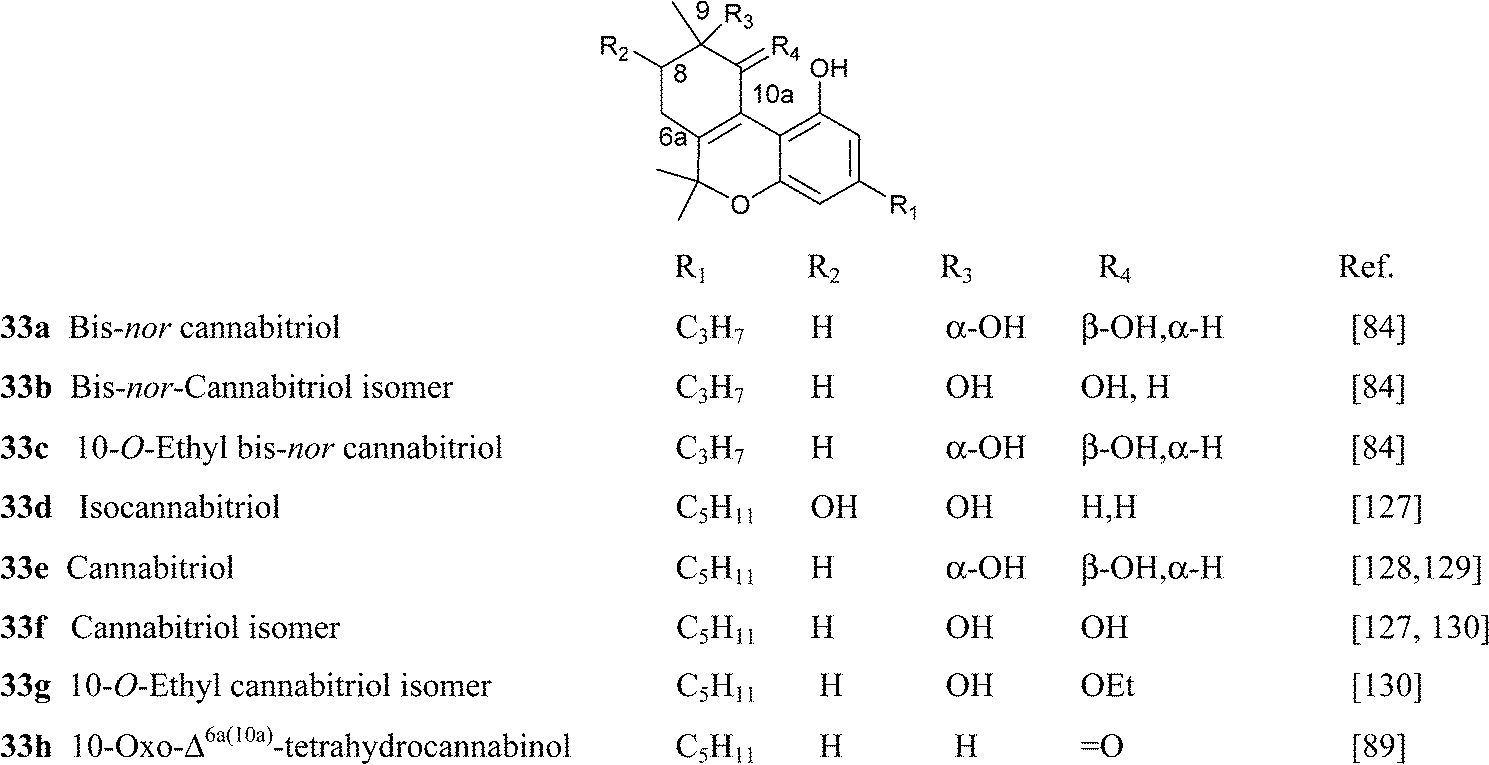 |
||
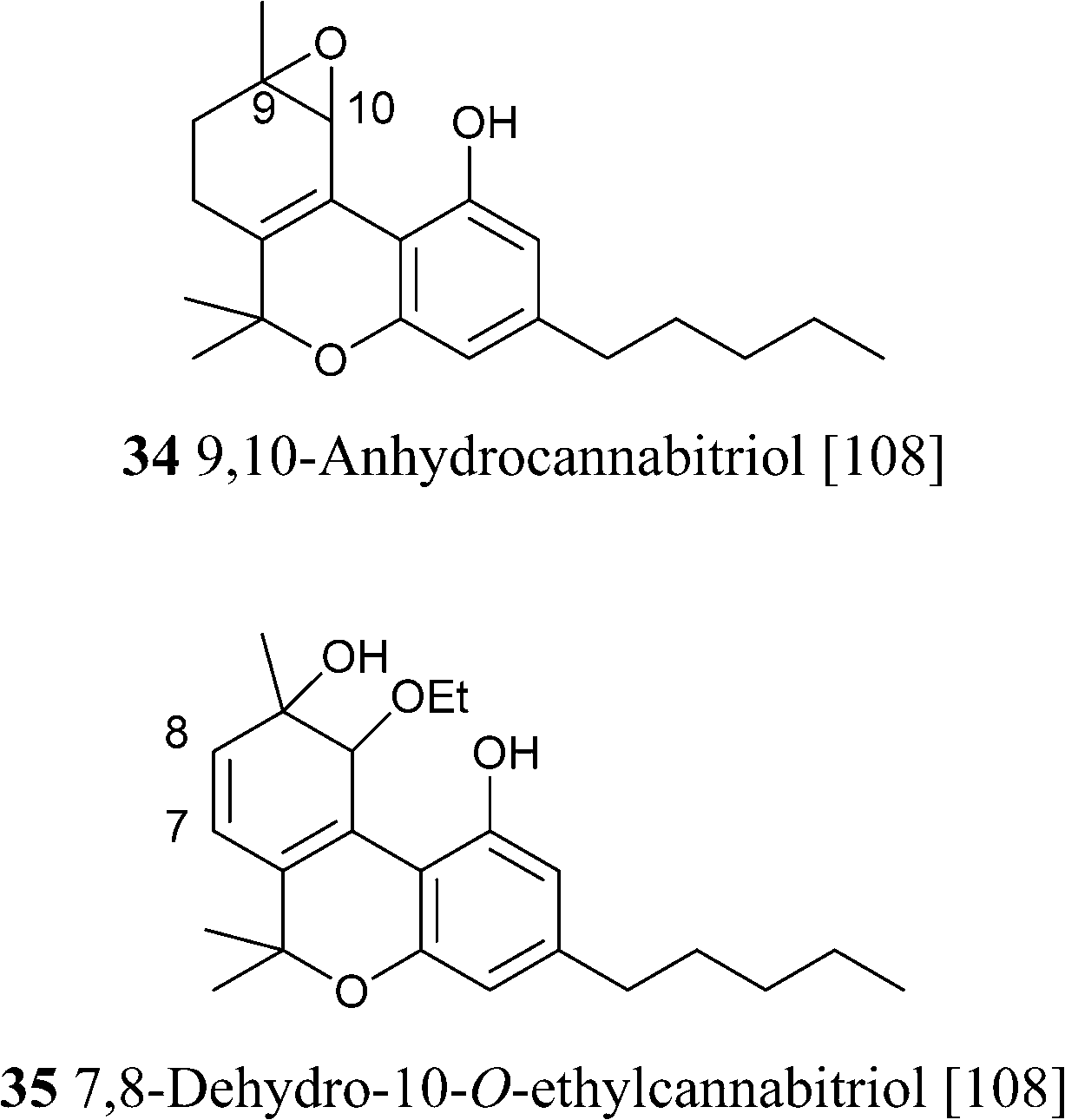 |
||
4.1.5.5. Isotetrahydrocannabinol-type compounds. Compounds from this class originate from CBD-type phytocannabinoids by protonation of the endocyclic double bond and quenching of the positive charge at C-1 by one of the two symmetrically disposed around C-3 (CBD numbering) phenolic hydroxyl of the resorcinyl moiety. While in THC-type phytocannabinoids the pyrane ring is linearly fused with the aromatic and the terpenyl moieities, in these compounds the junction is bridged. Both the stereochemical details and the biological profile of these compounds are still largely unknown.
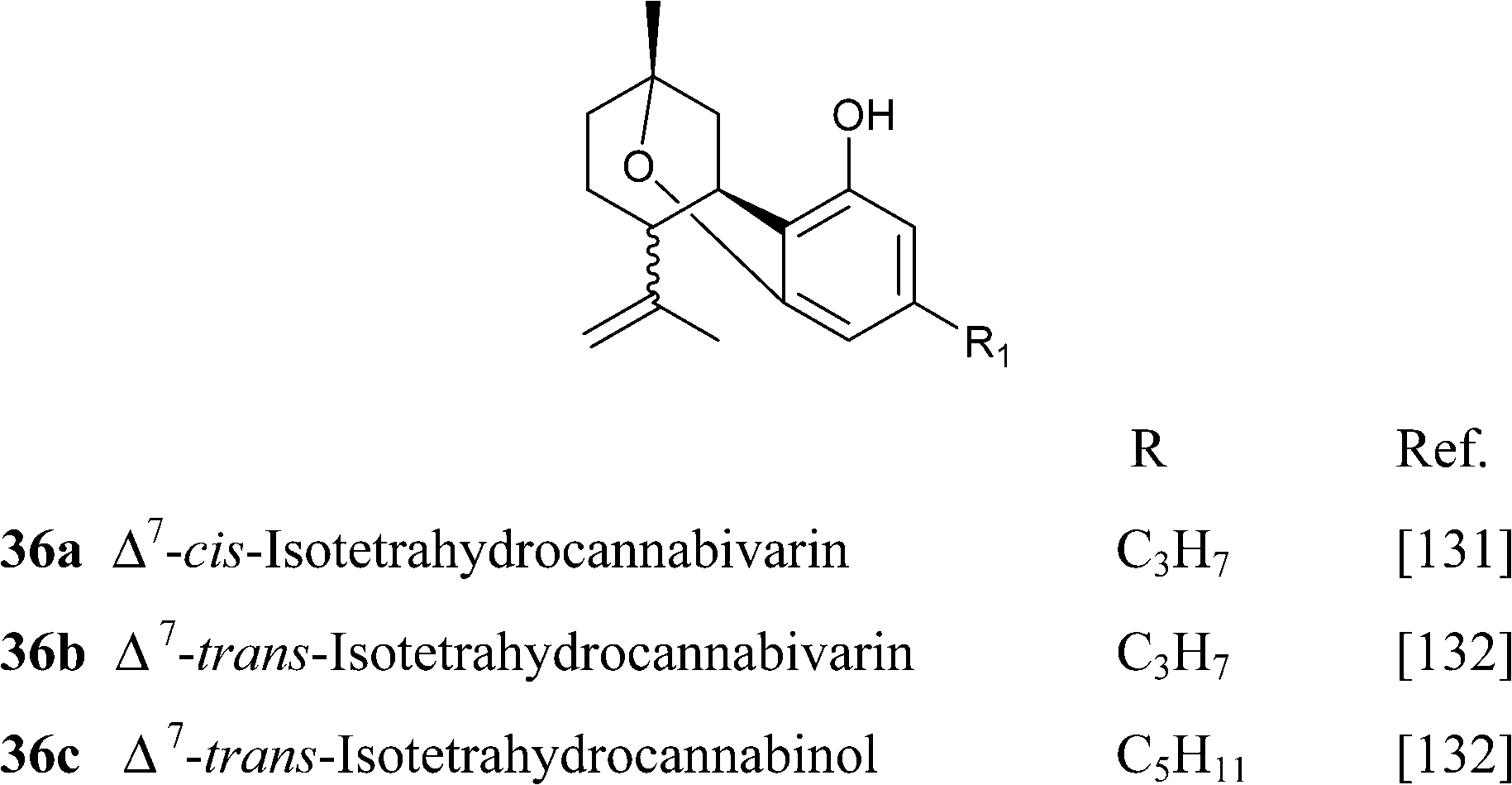 |
||
 |
||
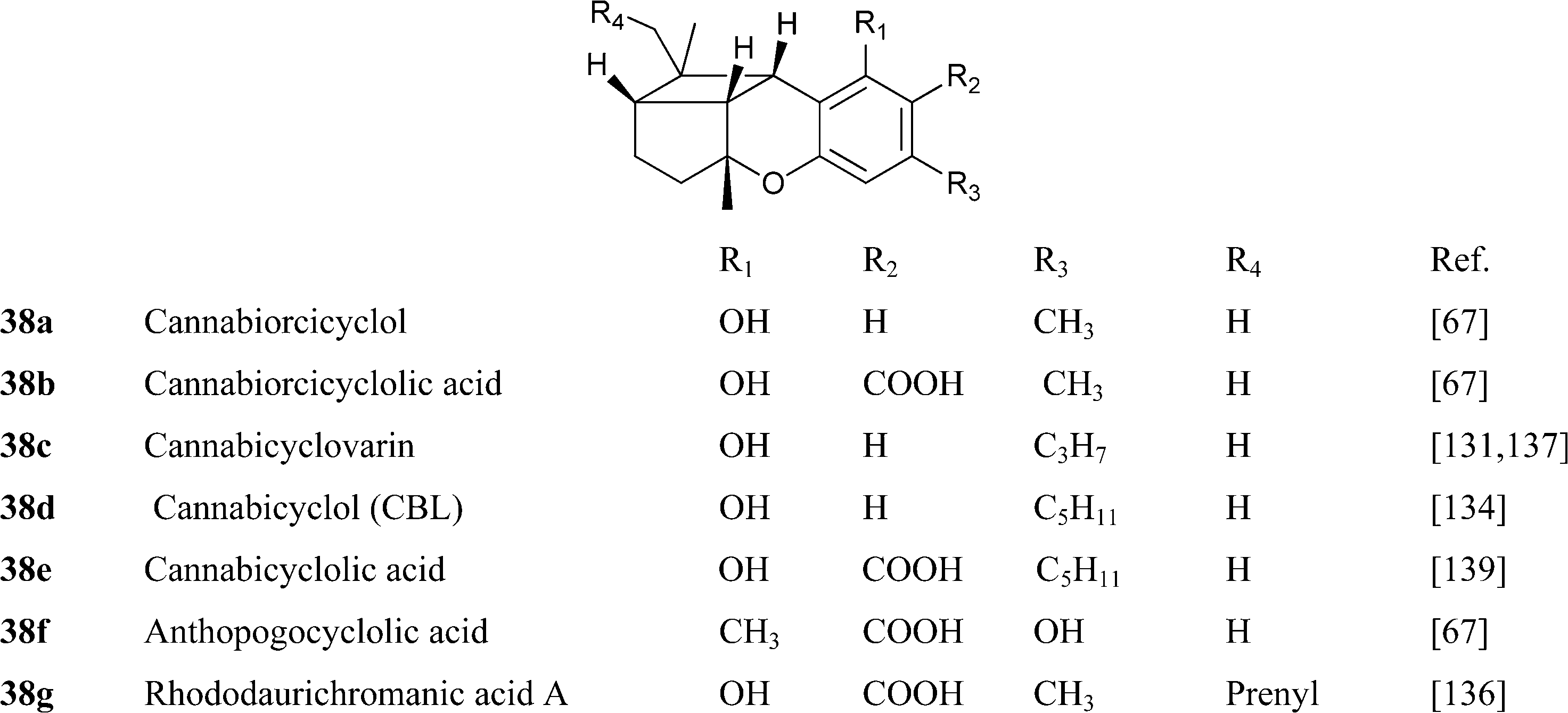 |
||
Two prenylogues analogues of CBE from the orcinoid series (ferrugienes A and B, 39f and 39g) have been isolated from the Alpine rhododendron (Rhododendron ferrugineumL.).74
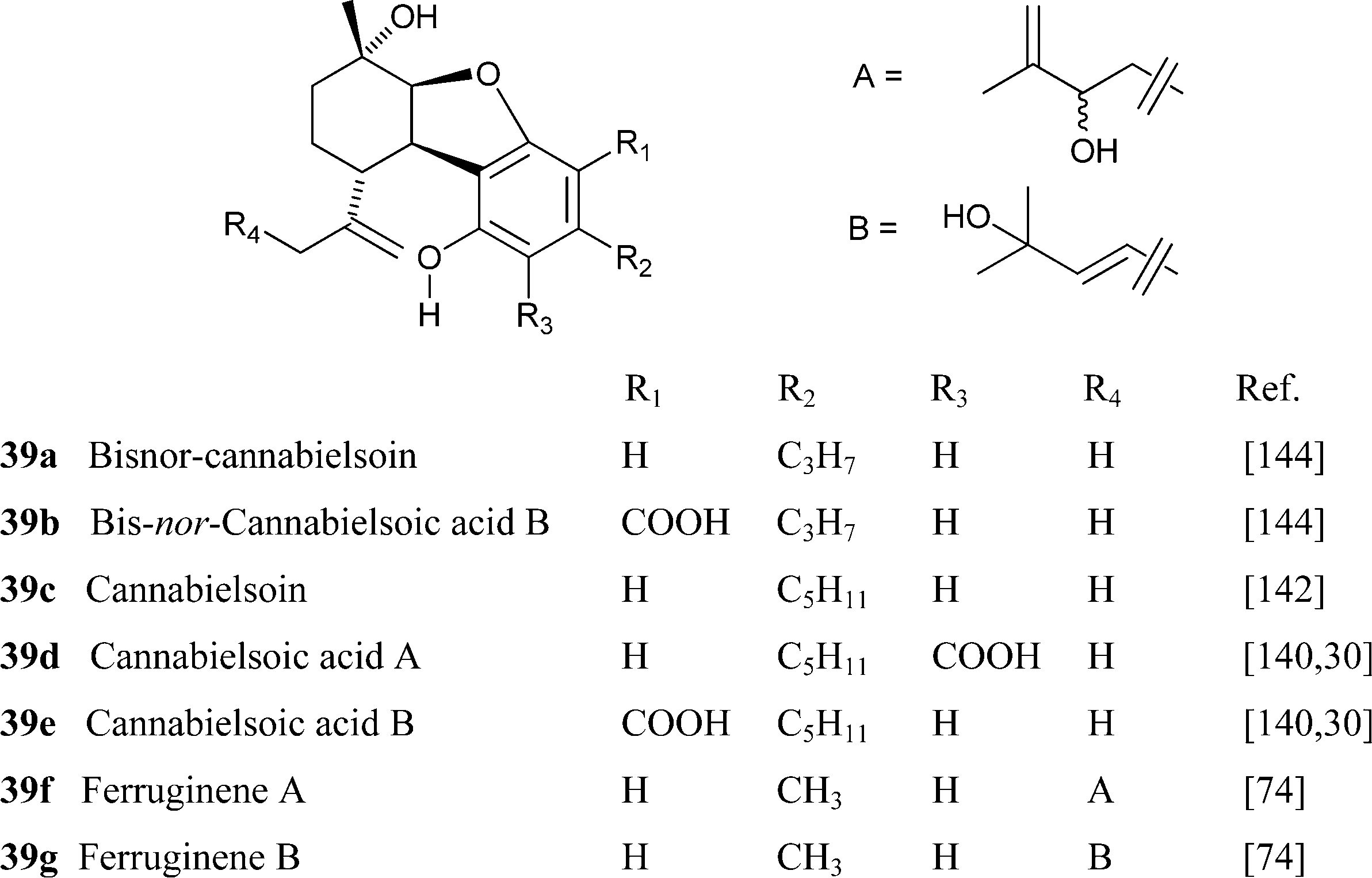 |
||
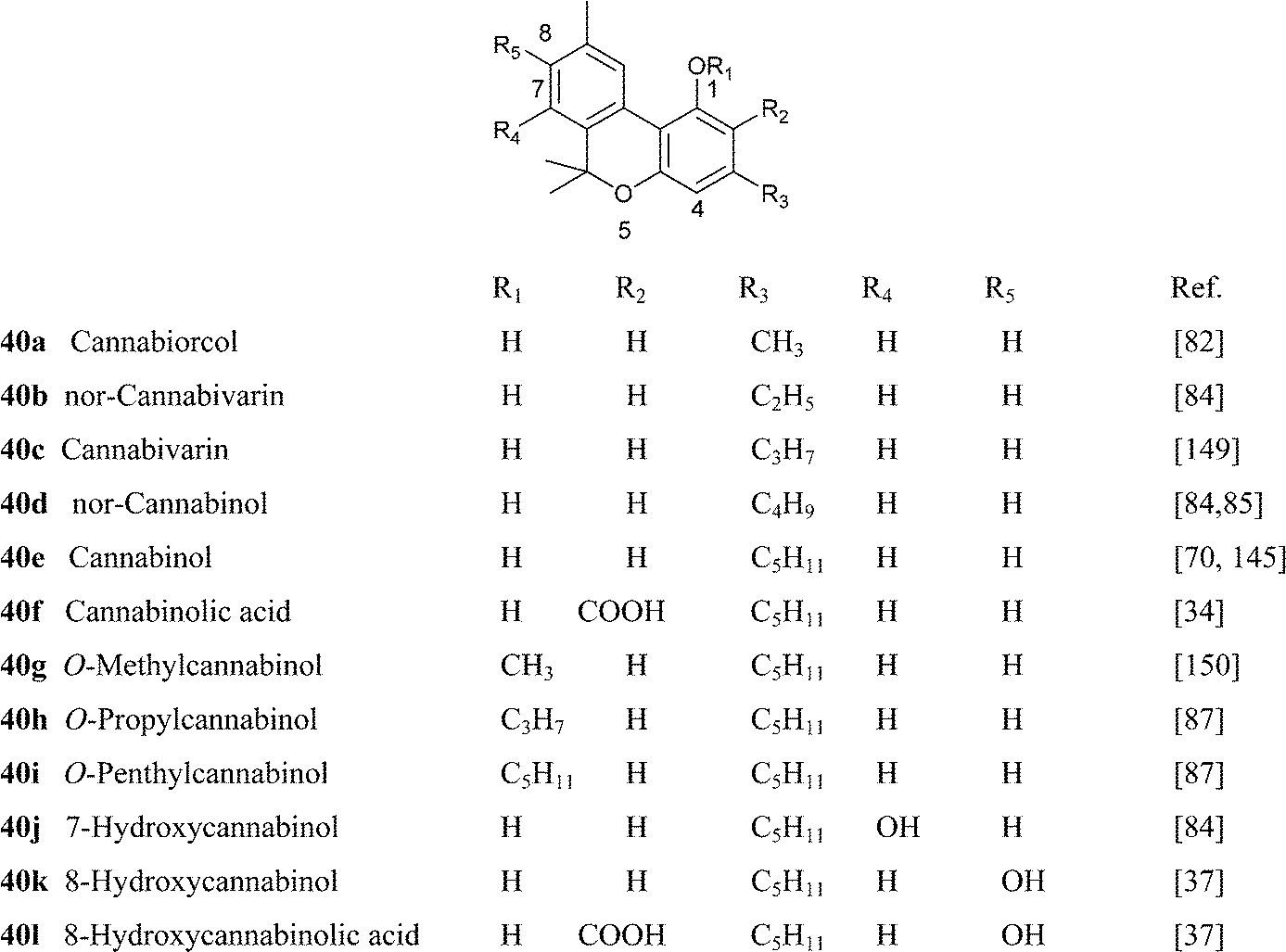
Cannabinol has only weak affinity for CB1 and CB2, ca. 10% of the one of THC.149 nor-Cannabivarin (40b), the only phytocannabinoid with an ethyl side chain, and nor-CBN (4d) were isolated from an historical bottle of cannabis tincture dating from the first half of the 19th century and prepared from an Indian sample of cannabis resin.84 The presence of phytocannabinoids with an even number of carbons could be typical of cannabis samples of that origin but, surprisingly, there are no modern studies on the diversity of cannabis in India.
 |
||
 |
||
 |
||
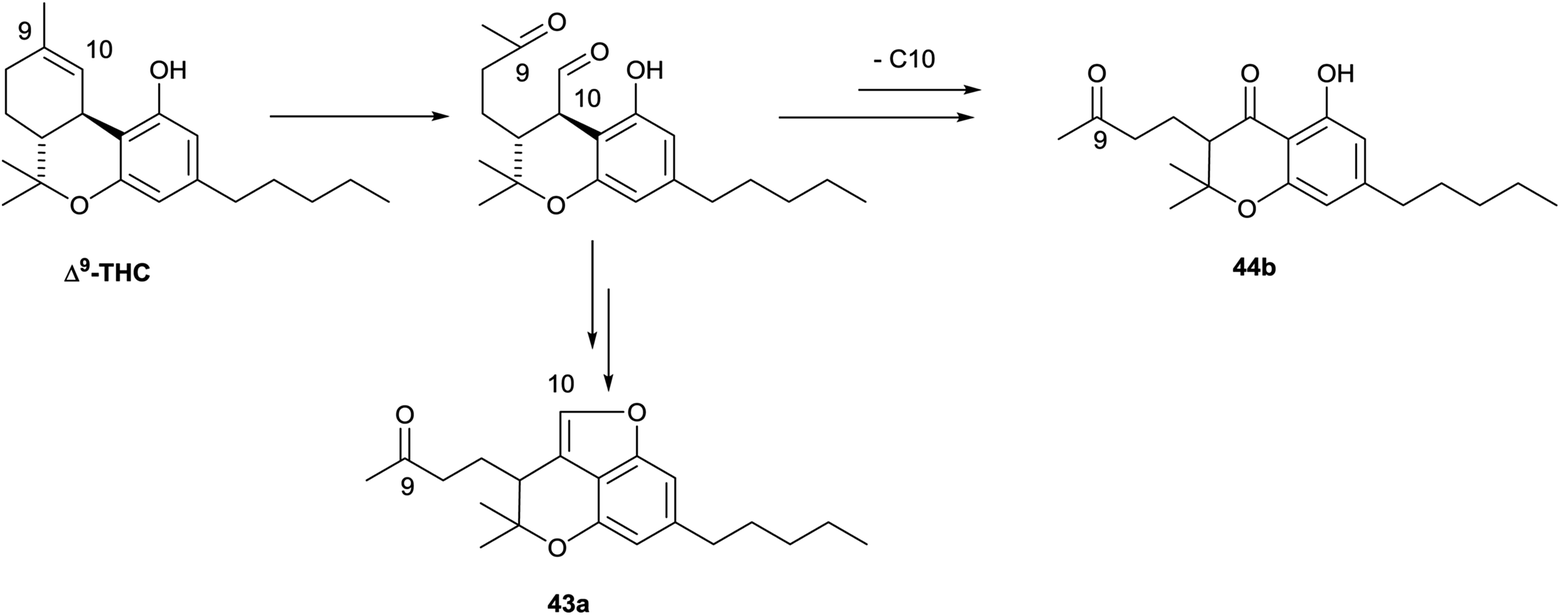 |
||
| Scheme 10 Oxidative degradation of the endocyclic double bond of Δ9-THC. | ||
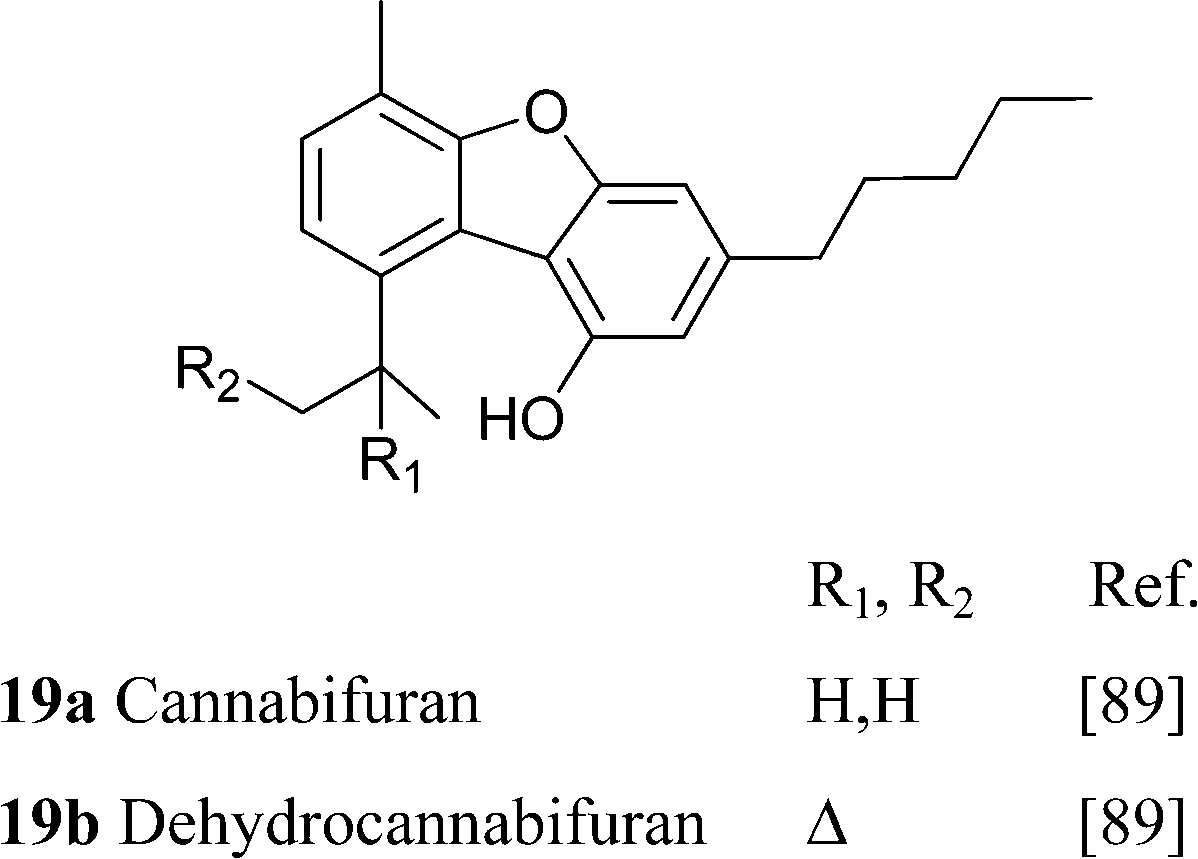
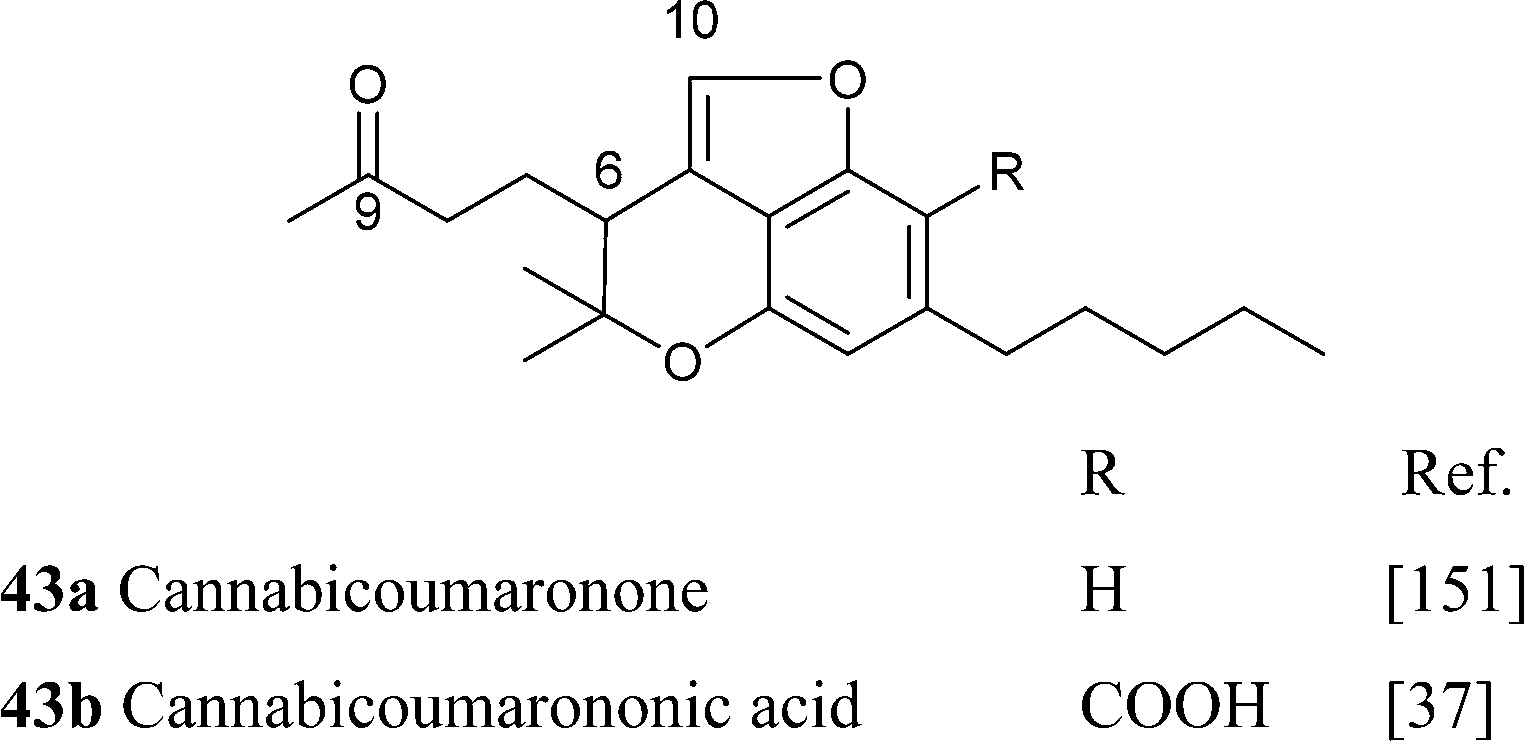
Further oxidative degradation of the furane moiety of cannabicoumaronone leads to cannabichromanones, a class of seco-10 norcannabinoids (Scheme 10). Cannabichromanone itself was isolated from a degraded sample of hashish having as major constituent CBN,89 and these compounds might well have a non-enzymatic origin.
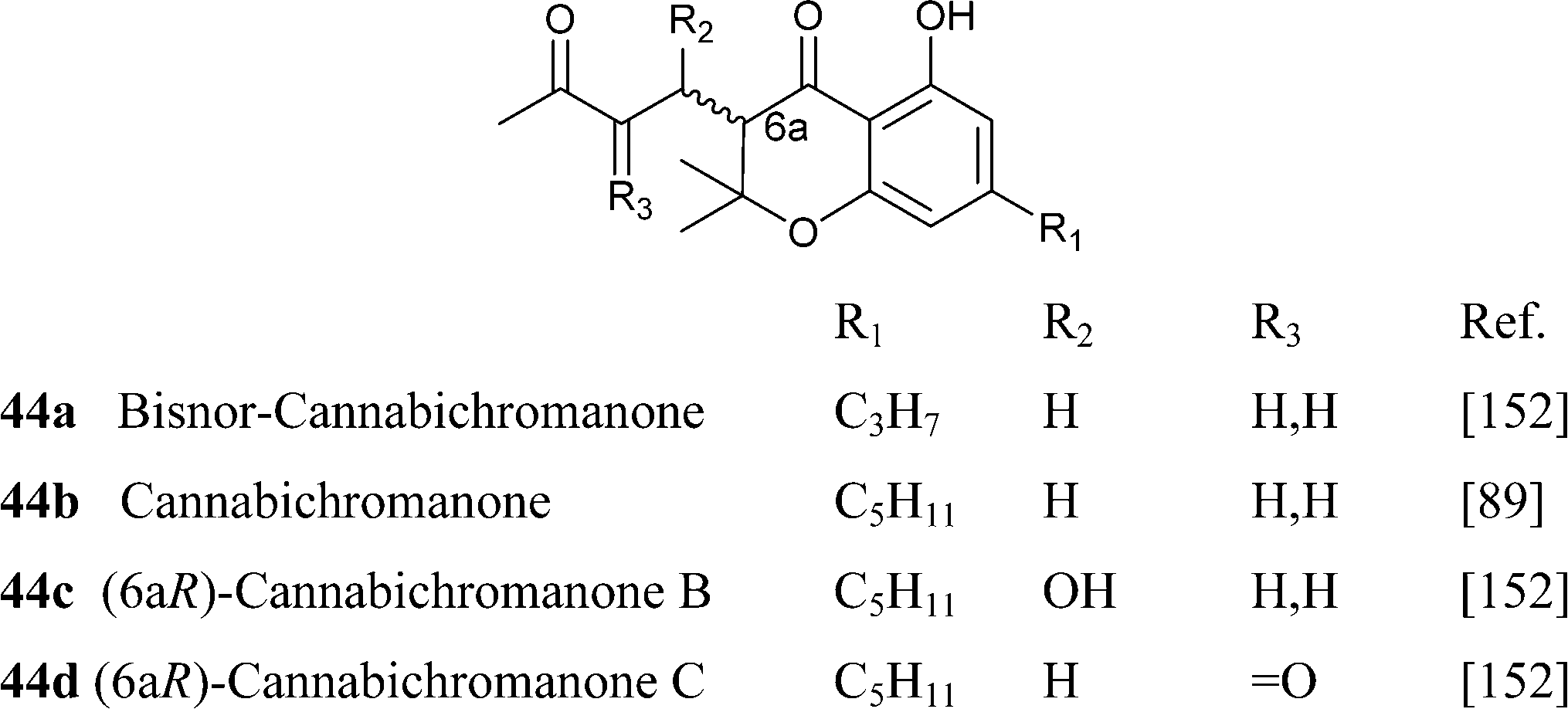 |
||
 |
||
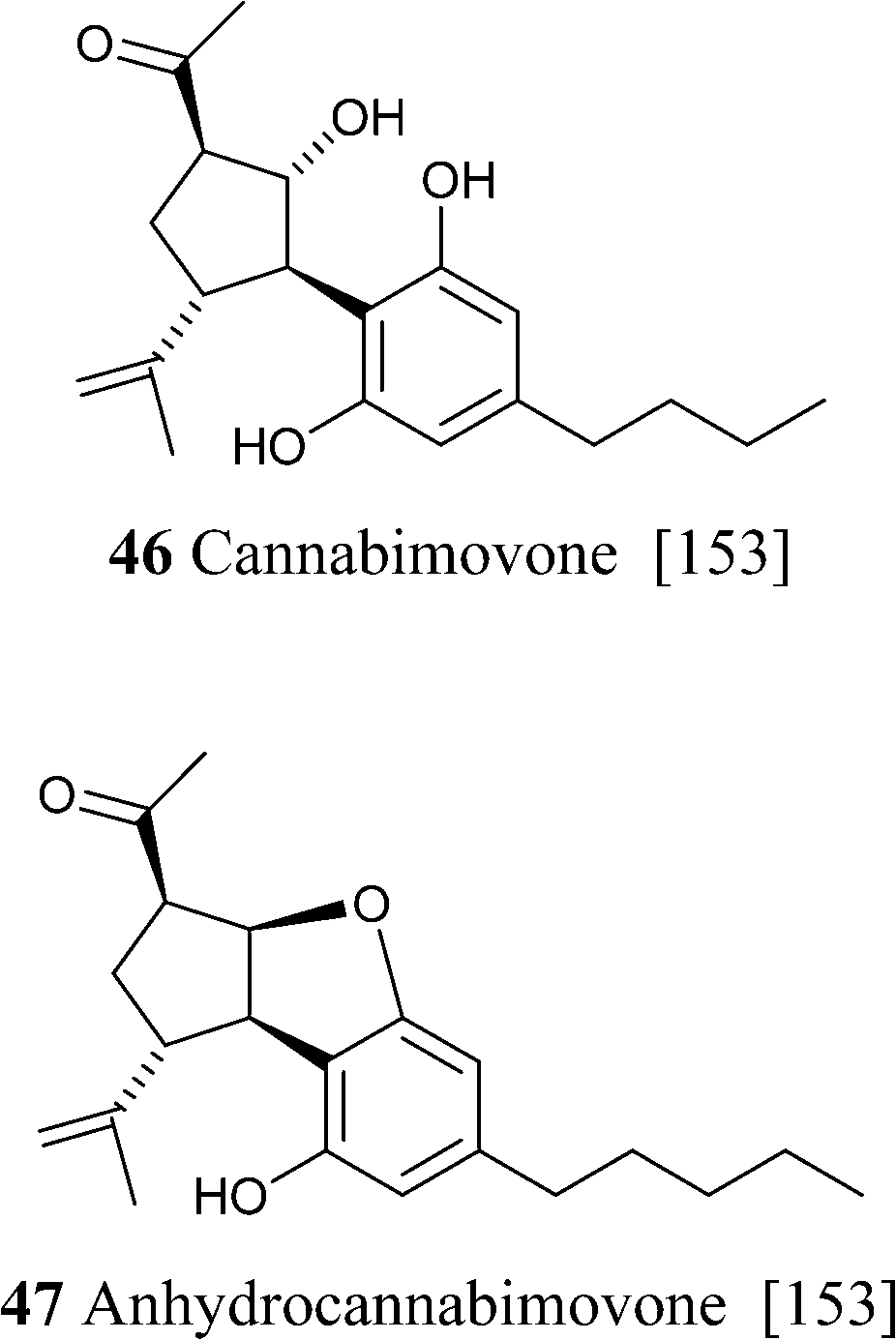
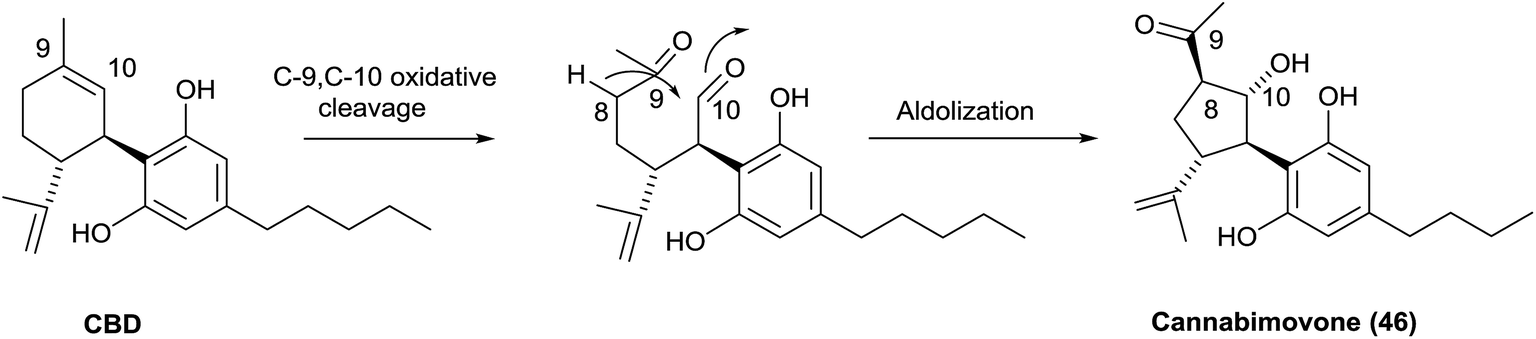
Cannabimovone (46) is formally the result of the oxidative fragmentation of the endocyclic bond of CBD followed by intramolecular aldolization (Scheme 11)153 Interestingly, attempt to mimic this biogenetic relationship with CBD failed to deliver the natural products, affording instead the oxy-Michael adduct of its crotonized version (anhydrocannabimovone, 47).153 While cannabimovone showed little affinity for CB1 or CB2, anhydrocannabimovone activated both CB1 and CB2 with a Ki of ca. 100 nM.153 The configuration of the oxygen bridge of anhydrocannabimovone was revised during the total synthesis of cannabimovone.154
 |
||
 |
||
| Scheme 11 Oxidative degradation of the endocyclic double bond of CBD to cannabimovone (46). | ||
4.2 β-Aralkyl type phytocannabinoids (phytocannabinoid-like compounds, bibenzyl cannabinoids, stiryl cannabinoids)
Because of the derivation from an aromatic starter, in these compounds a β-aralkyl residue replaces the alkyl group of cannabis phytocannabinoids, while the connectivity (but not always the configuration) of the isoprenyl moiety closely mimics the one of the cannabis products, overall resulting in similarity with the major phytocannabinoid chemotypes (CBG, CBC, THC). On the other hand, O-methylation of the resorcinyl moiety is rare within alkyl phtytocannabinoids, but is instead common in compounds from the β-aralkyl series, as are oxidative modifications of the isoprenyl residue, especially in compounds from the abnormal series.
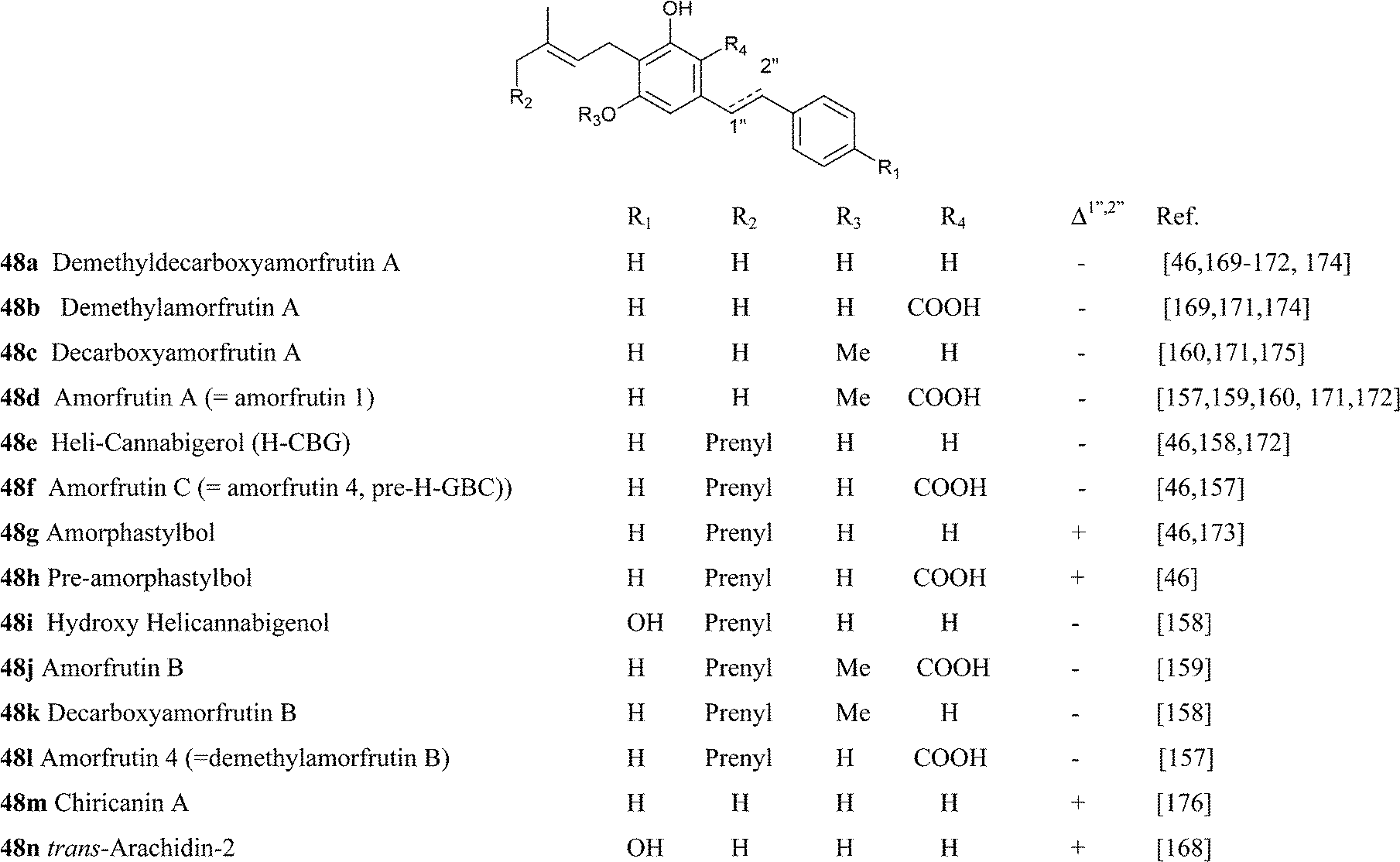
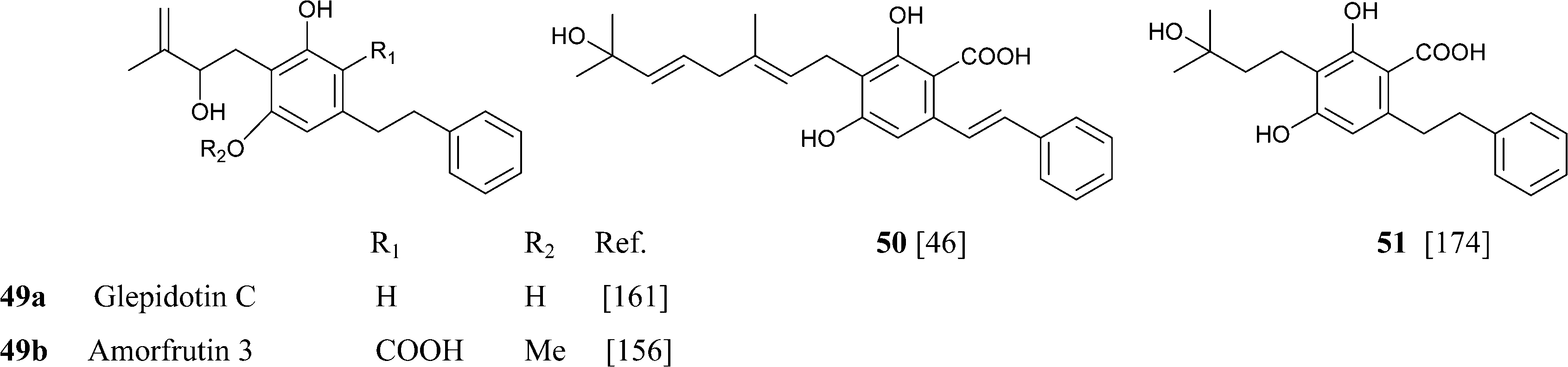
Amorfrutins were originally characterized as anti-bacterial agents,159 but interest was re-kindled by the discovery that amorfrutin B (48j) is a powerful ligand of PPARγ (Ki = 19 nM), showing remarkable insulin-sensitivity activity in vivo.157 The interaction of amorfrutins with PPARγ is basically different from the one of glitazones, since a crystallographic analysis has shown that amorfrutins bind PPARγ at the entry side and not at into the pocket of the ligand binding groove of this transcription factor.164 This finding underlies the observation that the amorfrutin-PPARγ complex associates to a distinct profile of proteins compared to the glitazone-PPARγ complexes, resulting in the selective activation of only of a subset of the genes under PPARγ control. The possibility therefore exists that the modulation of PPARγ by amorfrutins might not be associated to the side-effects typical of glitazones (fluid retention, weight gain, cardiovascular complication, bladder cancer), and animal studies have supported this suggestion.157 Amorfrutin B (48j) is the most powerful compound of the series in terms of PPARγ activation. Its superior activity compared to its demethyl derivative (amorfrutin 4, 48l) and it deprenyl derivative (amorfrutin A = amorfrutin 1, 48d) highlights the relevance of O-methylation and the oligomerization degree of the isoprenyl residue for superior potency. A second high-affinity target for amorfrutins was identified in the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH).165 Amorfrutins can inhibit both its activity and its translocation to the nucleus, a process involved in neuronal death, and hold therefore promise for the management, and possibly also the prevention, of neurodegenerative diseases. Several additional targets have been identified for amorfrutins, including the inhibition of NF-κB activity, the inhibition of iNOS, the corticotropin releasing factor-binding protein, the cysteine protease ATGB4, and the photoreceptor-specific nuclear receptor NR2E3.155,156 The multifaced profile of end-points makes it possible that amorfrutins could target, apart from diabetes, also a host of other conditions characterized by chronic inflammation, not unlike curcumin. For unclear reasons, amorfrutins and pre-cannabinoids from the phenethyl series are more resistant to decarboxylation compared to the alkyl phytocannabinoids.
Just like amorfrutins, also their analogues were isolated from taxonomically unrelated sources. Thus, the stiryl version of decarboxyamorfrutin C (amporphastilbol, 48g) was isolated from three leguminous Amorpha species (A. nana Nutt., A. fruticosa L., and A. canescens Pursh.),166 as well as from H. umbraculigerum, an asteraceous plant.46H. umbraculigerum also afforded its phenethyl analogue (48e), a compound first isolated from the liverwort Radula variabilis.158 In this context, the phytochemistry of H. umbraculigerum is very interesting, since this plant is not only the major natural source of cannabigerol in terms of isolation yield, but also produces its abnormal-, phenethyl- and stiryl- analogues, undoubtedly qualifying as the biogenetically most versatile source of phytocannabinoids known. Interestingly, also amorphastilbol was reported to bind PPARγ (as well as PPARα),167 but a direct comparison with amorfrutins has not yet been reported.
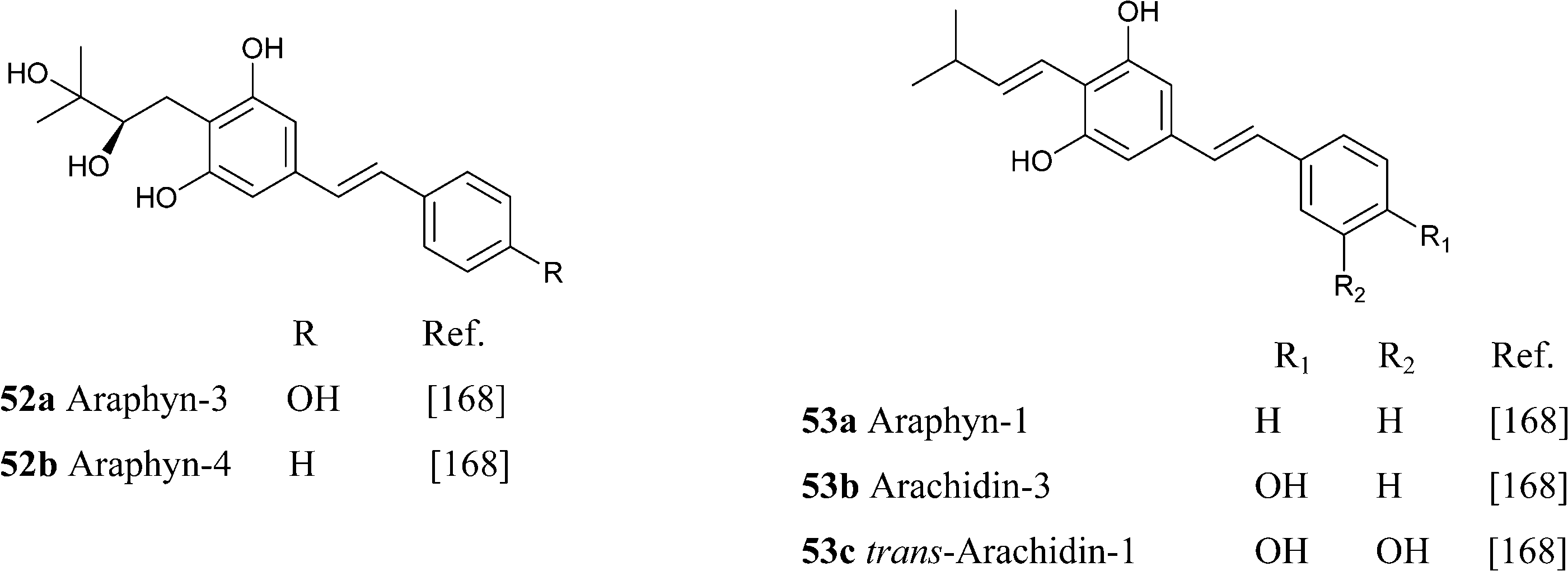
Amorfrutin-type compounds were also isolated from peanut (Arachis hypogea L.) seeds infected with an Aspergillus flavus fungal strain.168 Compounds 53a–c are characterized by a shift of the prenyl double bond in conjugation to the aromatic core, a rare feature in isoprenylated phenolics. These compounds (araphyns, arachidins) act as phytoalexins, helping the plant to resist fungal attack.
 |
||
 |
||
 |
||
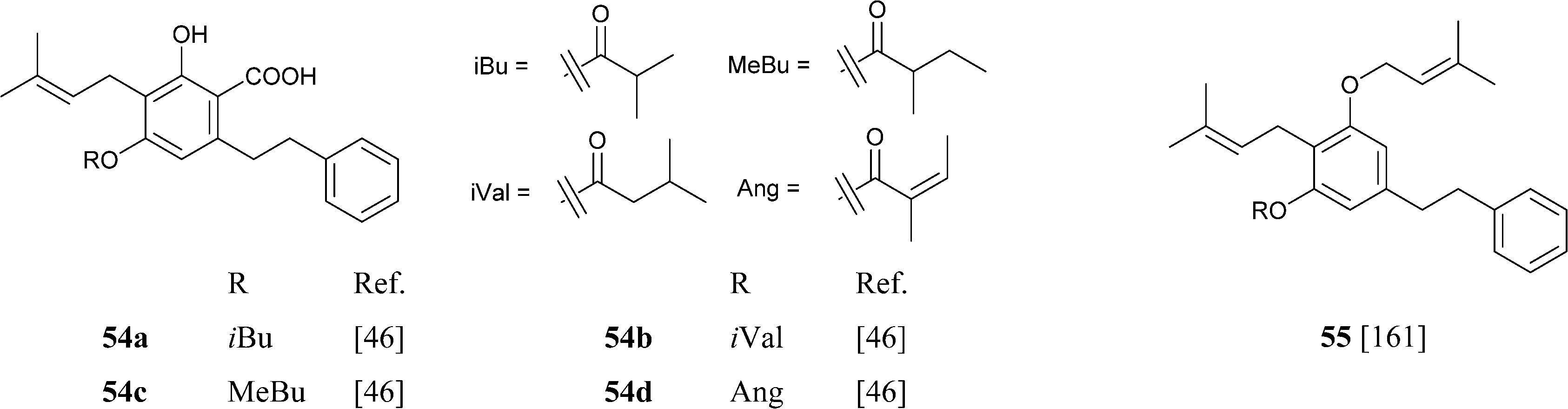
A unique feature of some phytocannabinoids from H. umbraculigerum is the esterification of the resorcinyl hydroxyl para to the carboxylate group, generally a site of methylation, with branched short-chain carboxylic acids.46 Within the phytocannabinoids from H. umbraculigerum, acylation is a selective feature of compounds from the phenethyl series with a prenyl residue, and was not observed in their stiryl and terpenyl analogues. O-Prenylation, along with meta-hydroxylation, has also been reported in a bibenzyl cannabinoid (55) from Glycyrrhiza lepidota.161
 |
||
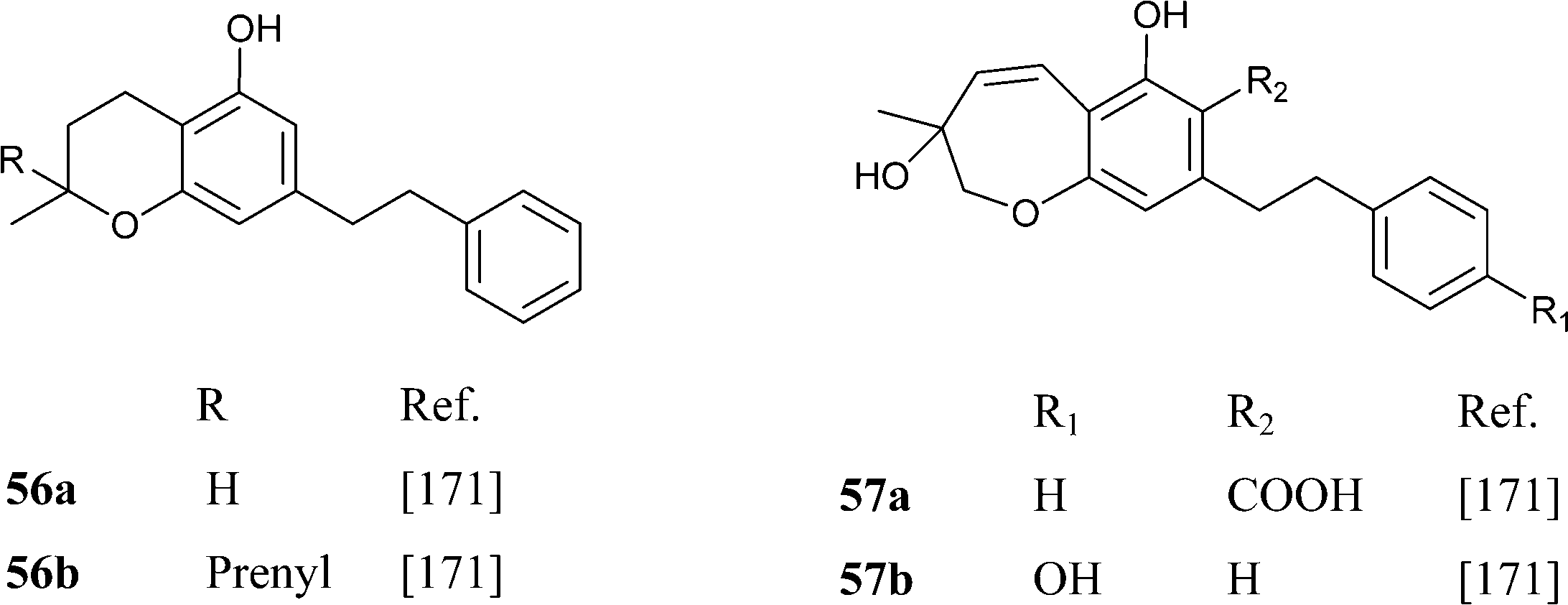
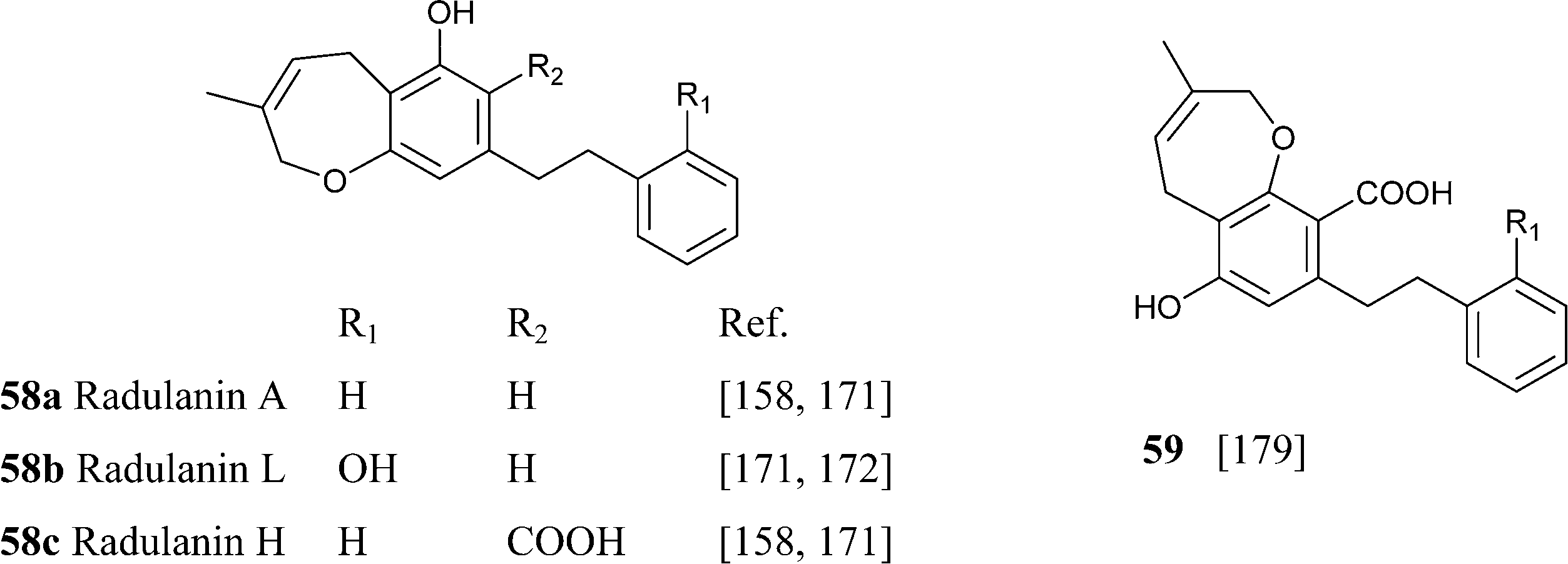
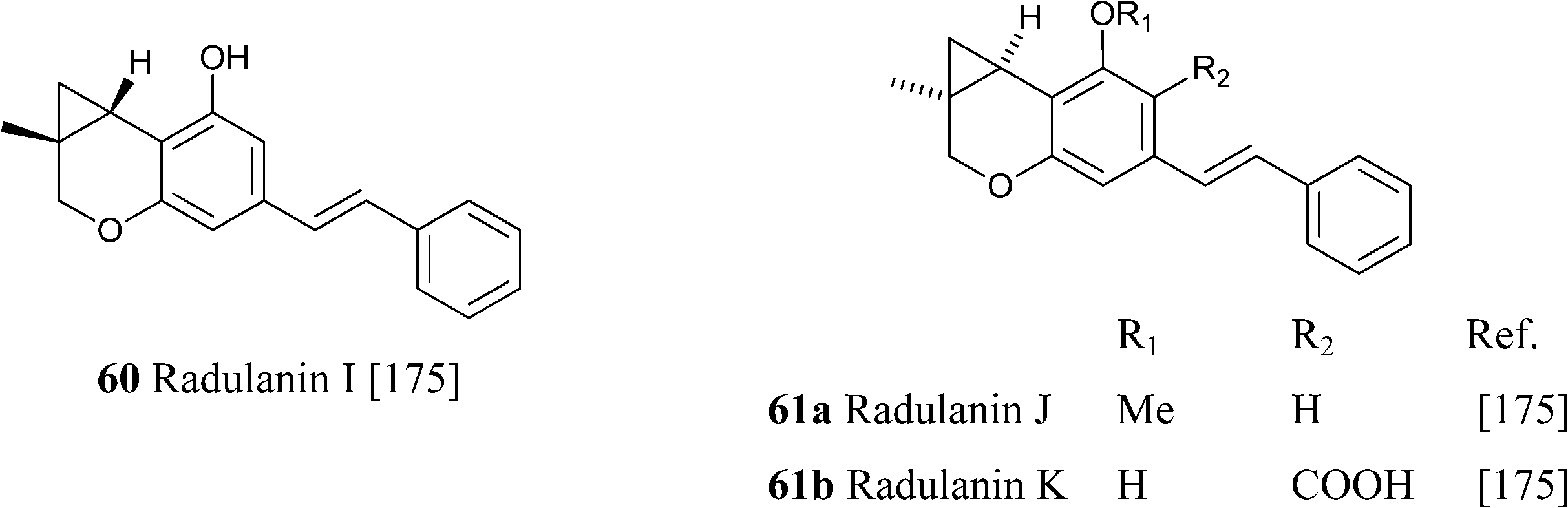
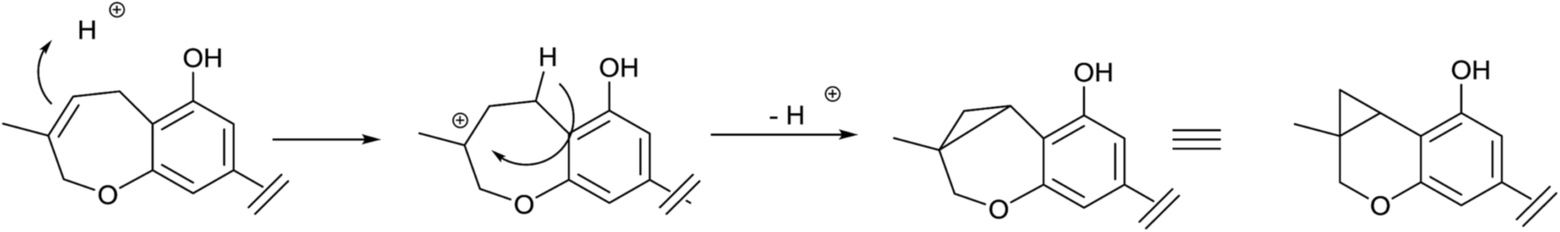
From liverwort of the Radula genus, stilbenic phytocannabinoids with an heterocyclized isoprenyl residue have been isolated. Apart from compounds resulting from the acidic cyclization of o-hydroxylated prenyl phenols, like compounds 56a,b and 57a,b, also compounds derived from the cyclization of ω-oxygenated precursors have been described.156Thus, compounds 57a–c are formally derived from the intramolecular opening of a terminal epoxide in a SN2 fashion (attack to the least substituted carbon) by the hydroxyl para to the carboxylate group. This 7-endo tet regiochemistry is unusual in isoprenylated phenolics, but its permitted by the Baldwin rules. A similar regiochemistry of intramolecular cyclization is involved in the generation 58a,c from their ω-hydroxylated precursors, with compound 58c being originally assigned the regio-isomeric structure 59. These compounds are, in turn, the precursor of the unusual cyclopropa-pyranes 60 and 61a,b, whose generation might involve the protonation of the oxepine double bond and then closure of a cyclopropane ring by loss of one of the benzylic protons (Scheme 12).
 |
||
 |
||
 |
||
 |
||
| Scheme 12 Possible biogenetic origin of the cyclopropapyranes 60,61a,b. | ||
The “taxonomy” of a series of chromanes from leguminous plants and liverworts is ambiguous. Biogenetically, they could be considered either as cyclized CBG-type compounds, derived from the cyclization of a prenyl (56a,b) or an ω-epoxyprenyl precursor (62a,b). Alternatively, as hydrogenated or hydrated CBC analogues. The CBG-type derivation seems more likely, and therefore they are included in this section.
 |
||
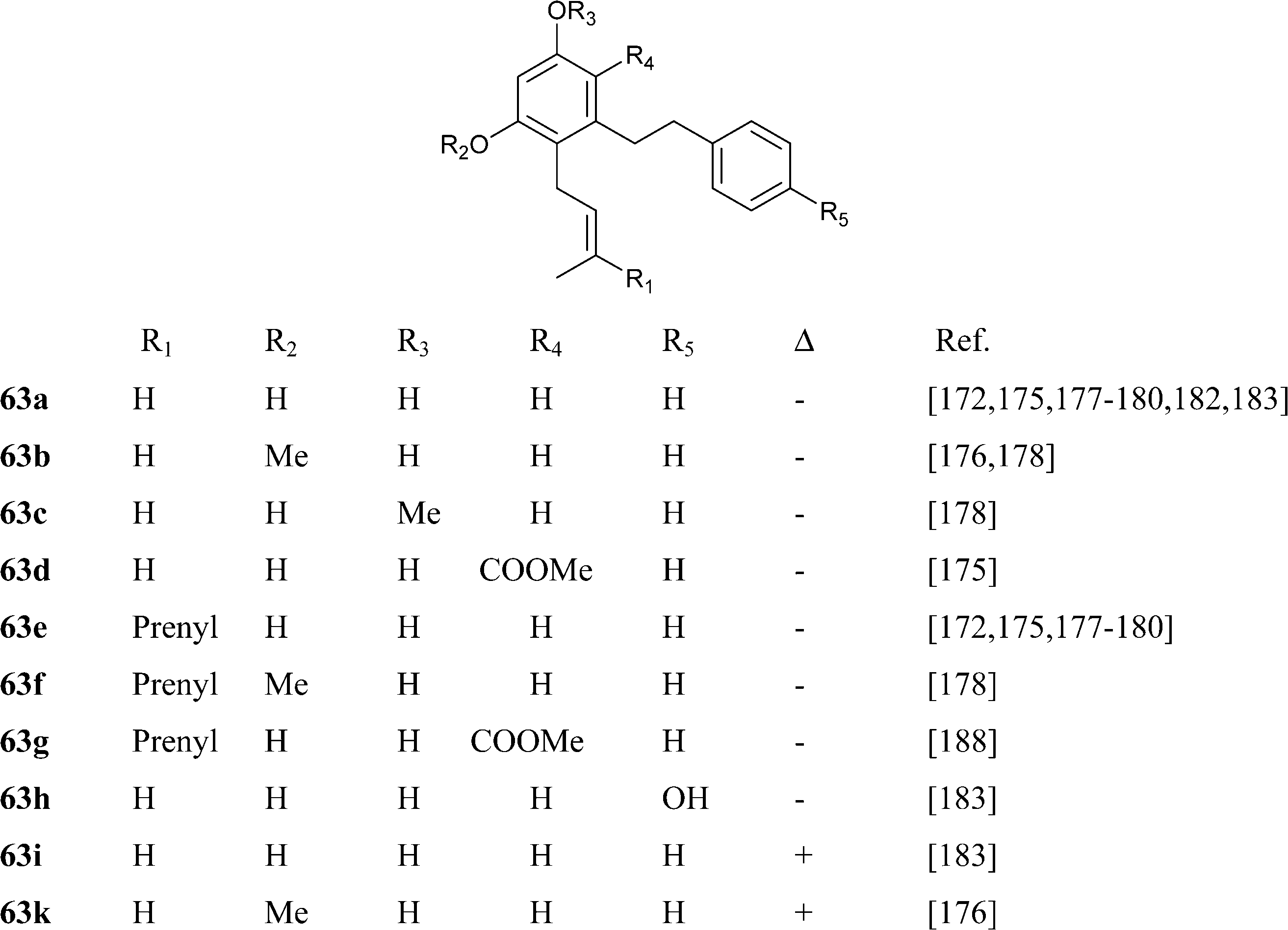
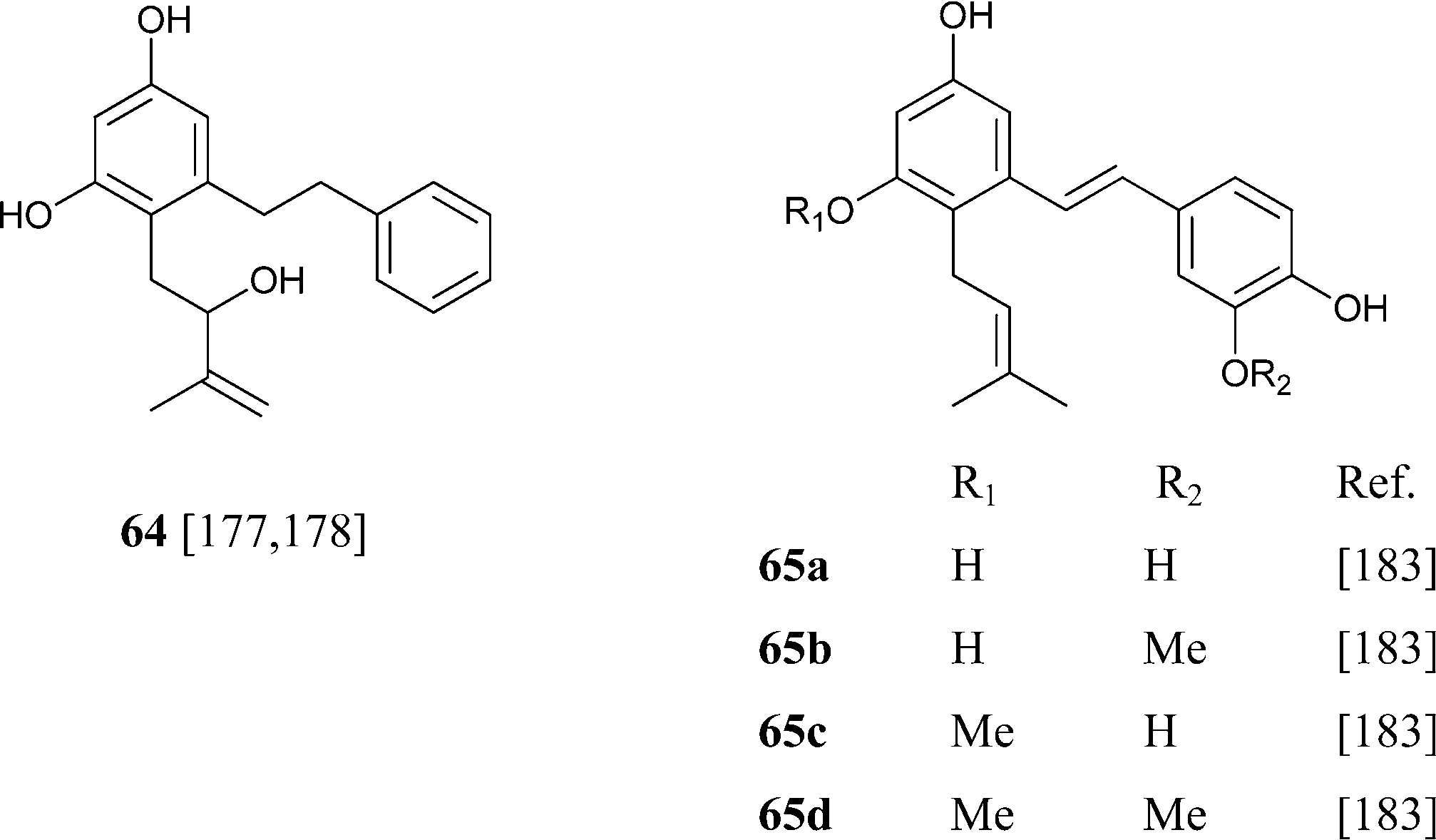
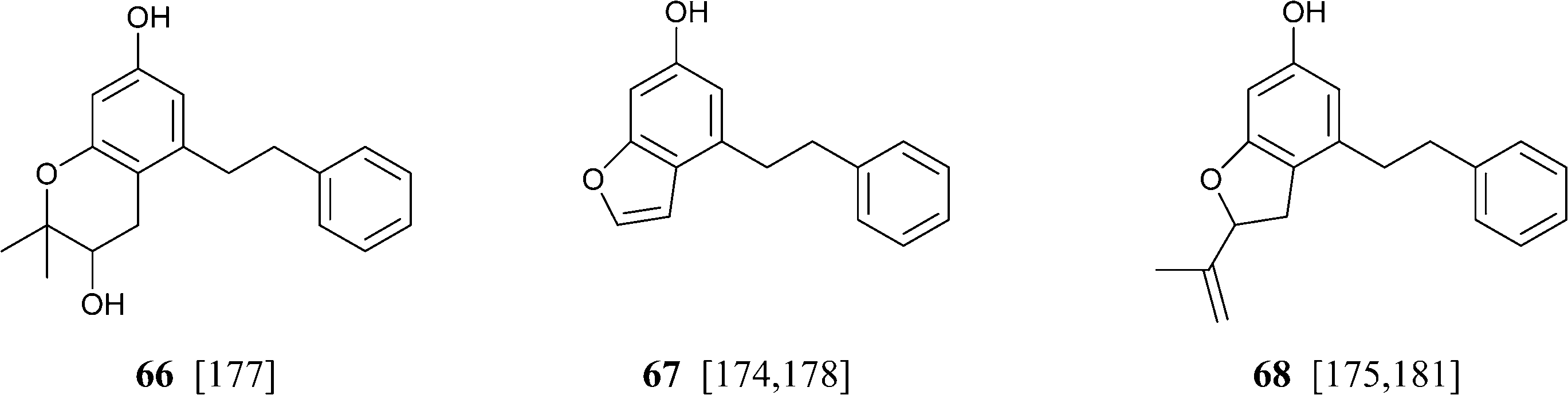
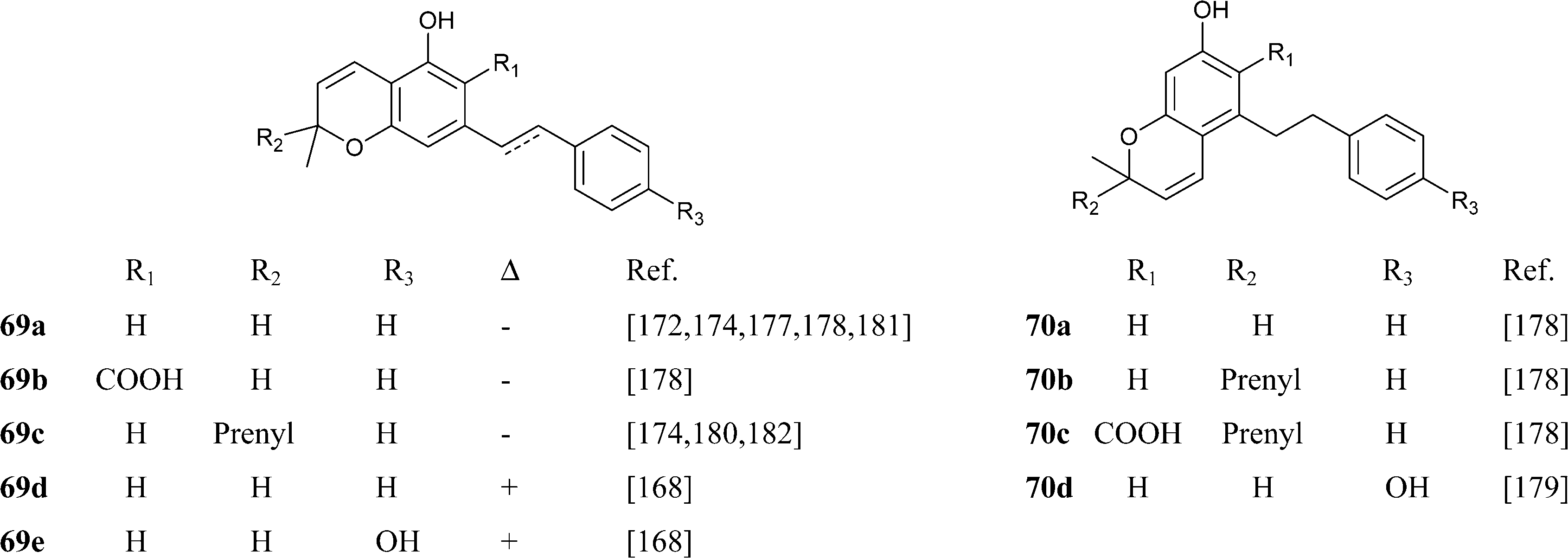
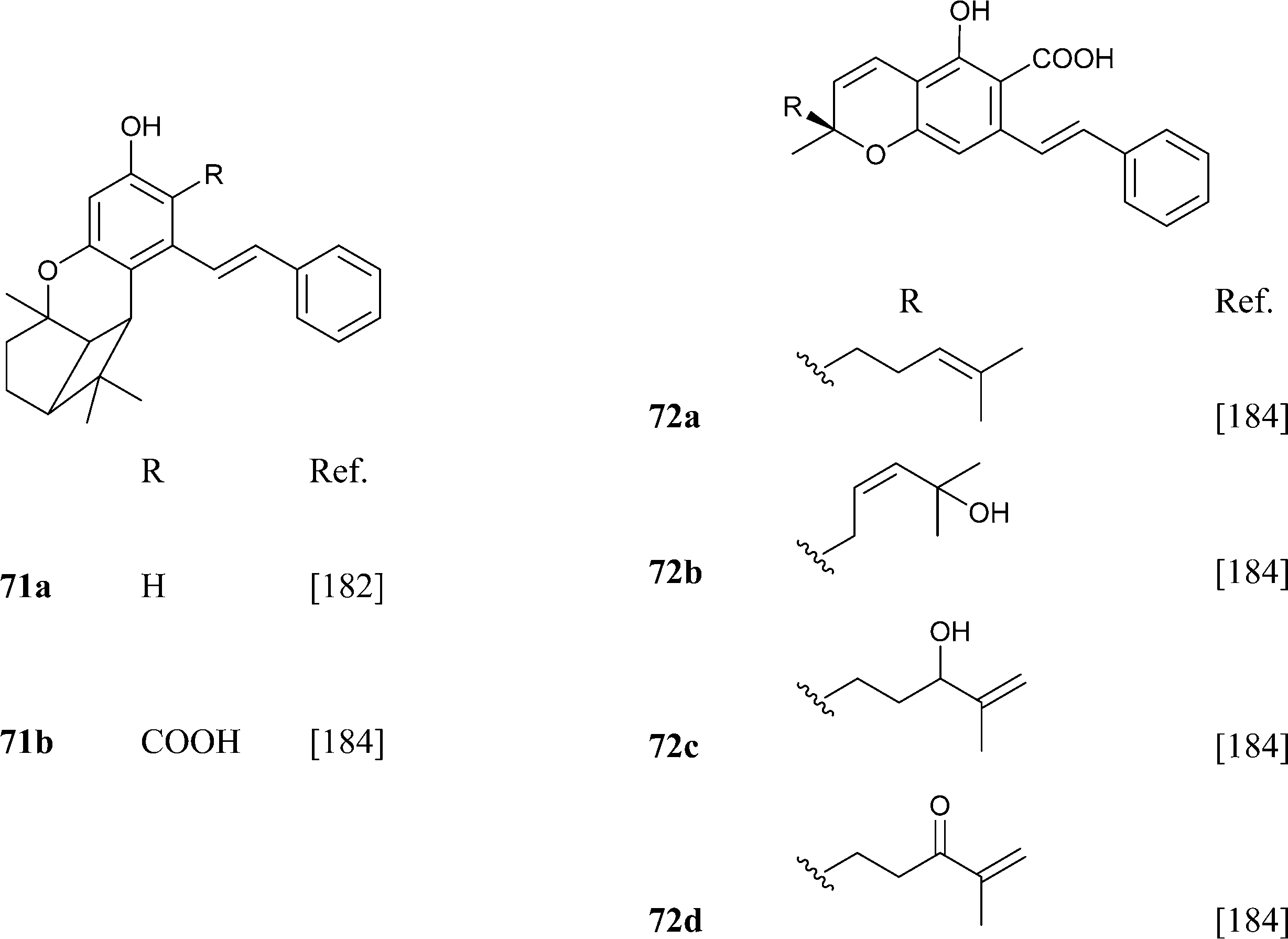
Abnormal phenethyl phytocannabinoids are widespread in liverworts from the genus Radula, where, like in R. variabilis177 and R. kojana,178 they can represent the major chemotype of bibenzyls, or even the only type of phytocannabinoids detected, as in R. voluta.179,180 Remarkably, R. perrottetii contains abnormal phytocannabinoids from the CBG and CBC series, and regular phytocannabinoids from the menthyl-type (THC series).181 The structural diversity of phenethyl abnormal phytocannabinoids closely parallels the one of their related regular phytocannabinoids (O-methylation, prenylation), but also “internal” hydroxylation of the prenyl residue has been reported, as in 64 and 65. The furan 67might derive from the degradation of the isopropyl-substituted dihydrobenzofuran derivative 68, as usual in the biogenesis of furanocoumarins from plants.
 |
||
 |
||
 |
||
 |
||
 |
||
 |
||
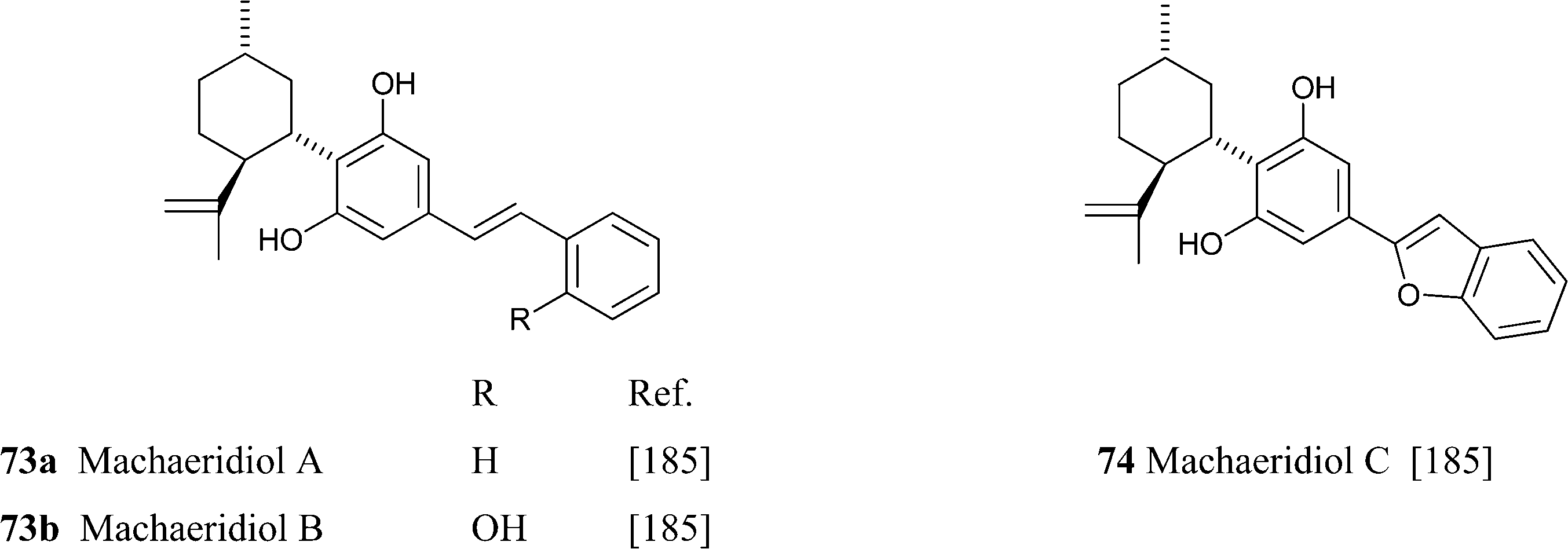
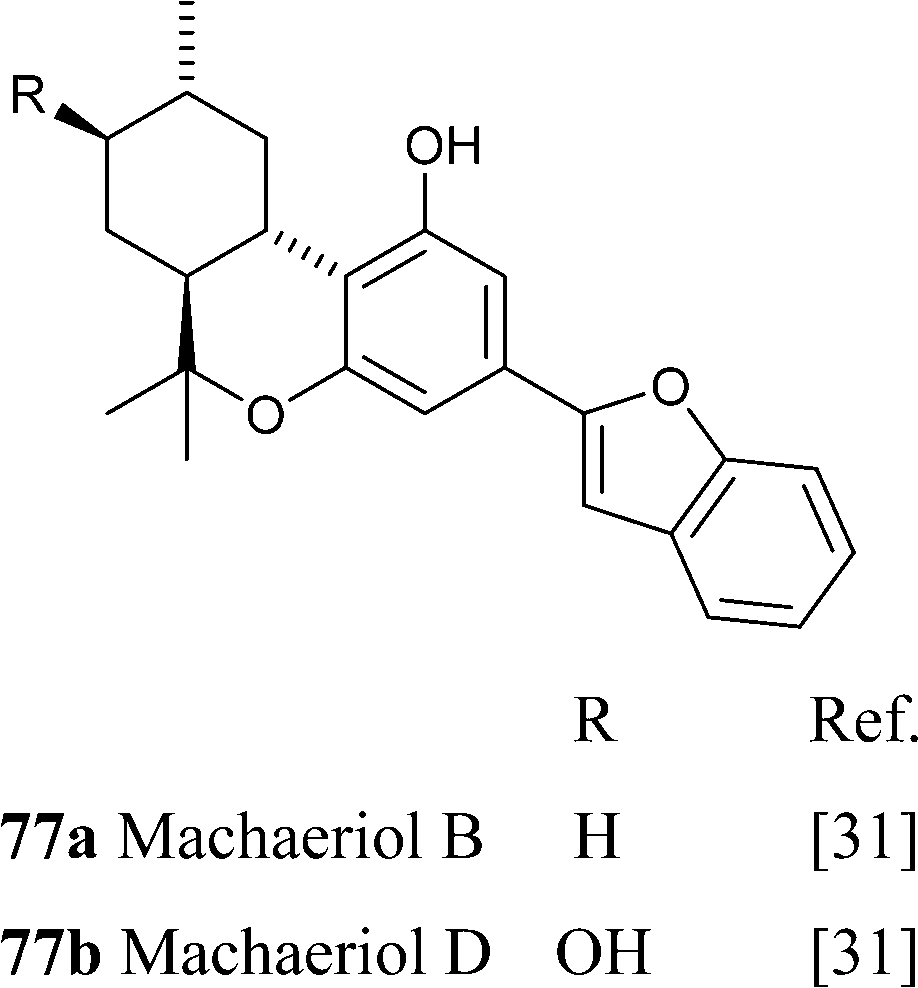
The macheridiol chemotype is similar to the one of CBD, with the β-aralkyl moiety declined in the stiryl (73a,b) and benzofuranyl (74) form. These compounds, as well as the THC analogues from the macheriol chomotype (see infra),186 were isolated from the stem bark of the Amazonian legumionous liana Macherium multiflorum Spruce.185 The pseudo-enantiomeric configuration at C-3 and C-4 compared to CBD was suggested by CD studies. Despite their similarity, the biological profile of machaeridiol is remarkably different, with machaeridiol B (73b) being an order of magnitude more potent of machaeridiol C (74) as an antimalarial agent.185
The occurrence in Nature of the phenethyl analogue of THC was predicted in 1986 by Crombie,187 an overlooked founder of cannabinoids (and not only this class of compounds) chemistry, based on the occurrence of the phenethyl analogue of CBG, the precursor of THC in cannabis, in lieverworts158,172 and in higher plants.46 Two years later, Crombie synthesized the phenethyl version of THC with the aim of investigating its presence in cannabis, but no information on its bioactivity was disclosed.188 While the phenethyl version of THC is still unknown as a natural product, its cis isomer [perrottettinen(e)] was isolated by Asakawa from the Japanese liverworth Radula perrottetii181 and from the New Zeeland liverworth Radula marginata,177 and by Becker from the Costa Rican liverwort Radula laxiramea,174 with the absolute configuration being confirmed by an enantioselective synthesis.189 Since cis-THC, a very minor cannabinoid in marijuana but almost equimolar with THC in fiber hemp, is not psychotropic,85 also perottetinene should not be so. On the other hand, detailed information on the biological profile of the various isomers of THC has never been published, and the biological profile of perrottettinene is unknown, or, at least, it has not been reported in the mainstream literature, despite undocumented claims on its psychotropic properties that circulate on the web.190 It is remarkable that the enormous efforts of exploration of the biological space around the THC chemotype and the critical role of the C-3 substituent on bioactivity, the “hint” suggested by Nature with the existence of phenethyl versions of the pentyl cannabinoids of Cannabis has been so far overlooked. Since cannabinoids have additional targets to the psychotropic CB1receptor, the exploration around the perrottetinene chemotype seems well worth pursuing.
Machaeriols A and B from the Amazonian liana Machaerium multiflorum Spruce are analogues of trans-dihydroTHC,186 but show an enantiomeric configuration at the ring junction, as shown by CD studies and enantioselective total syntheses.191–193 It is not known if machaeriols bind CB1 and are psychotropic.
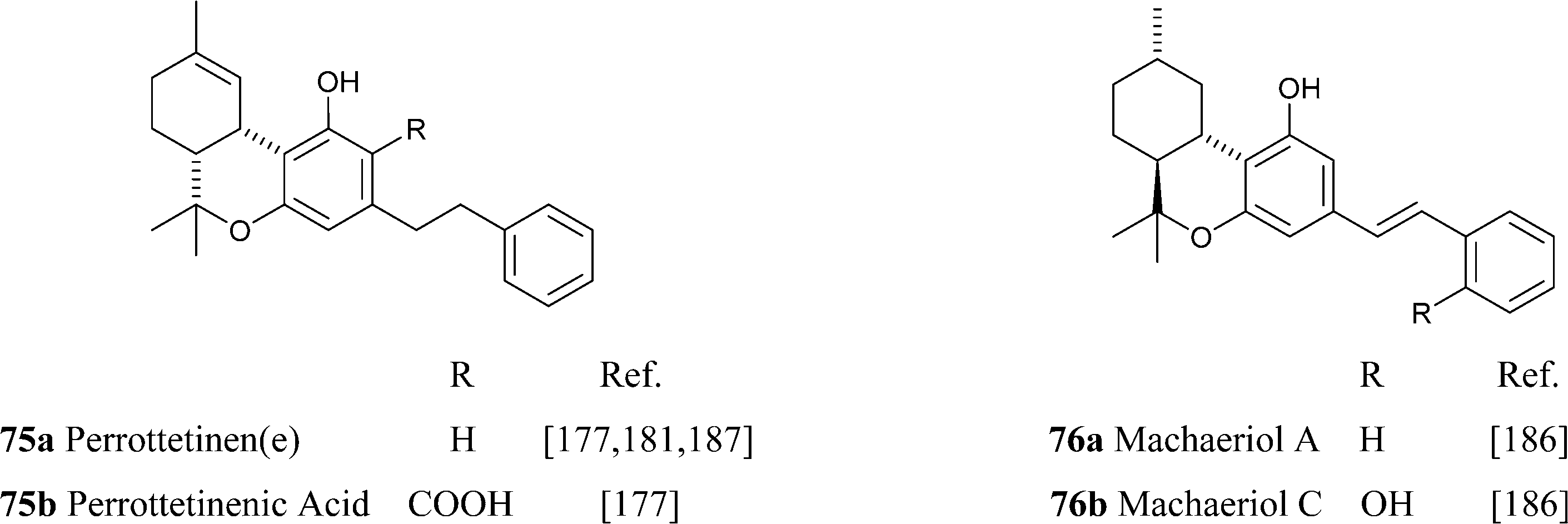 |
||
 |
||
 |
||
 |
||
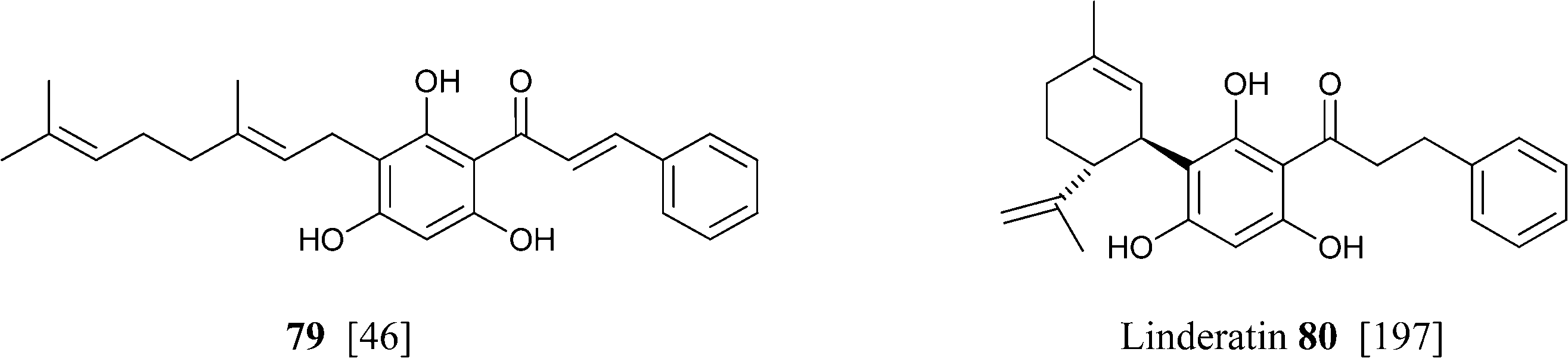
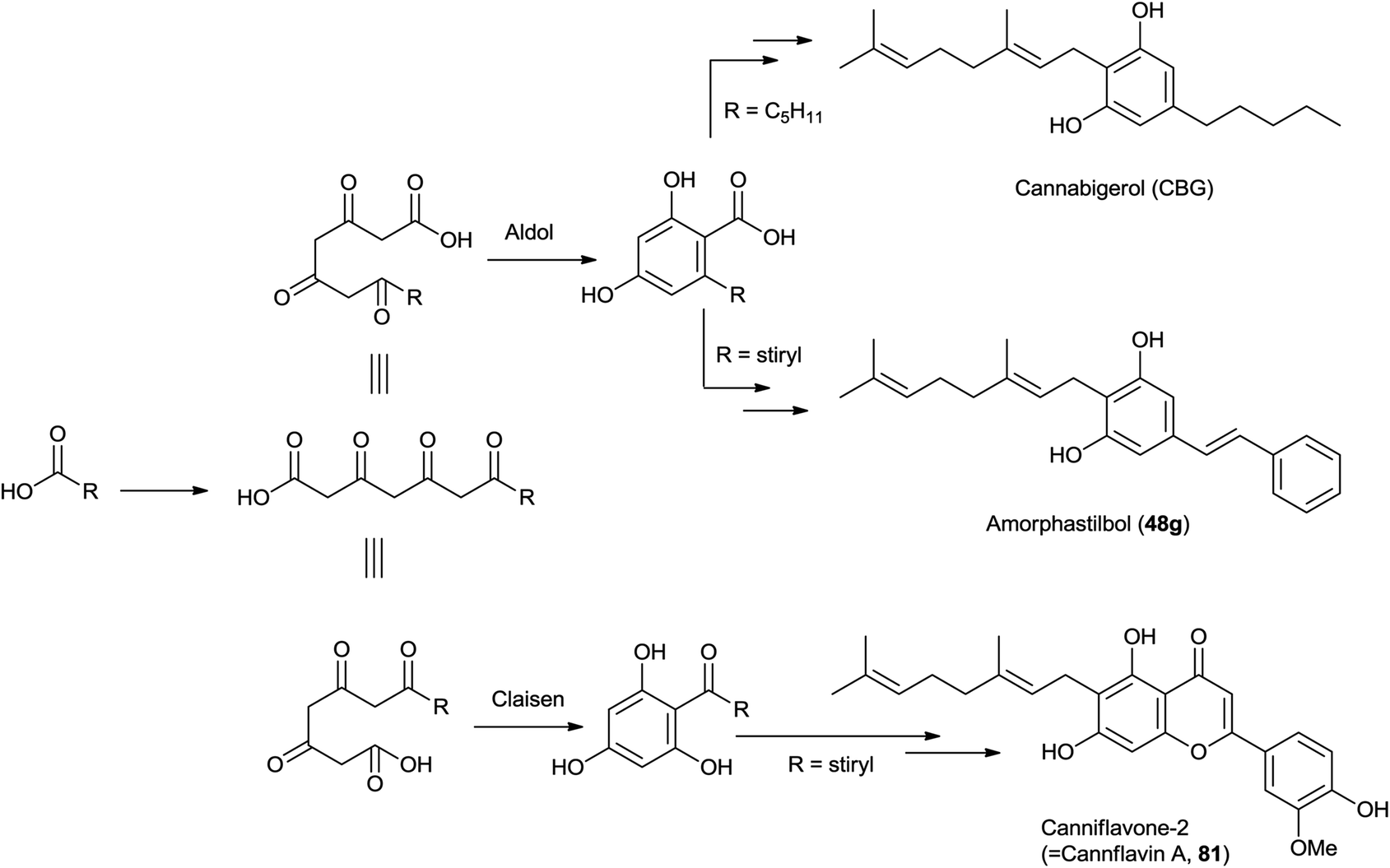
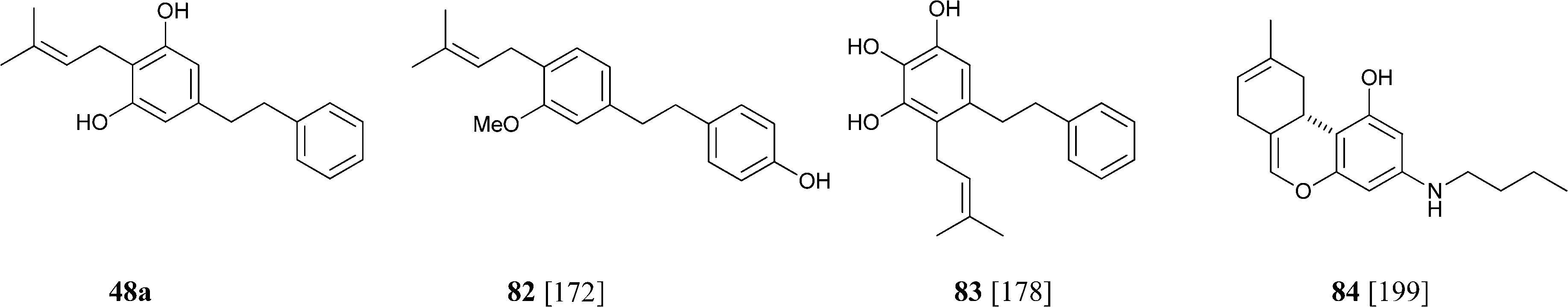
Similar considerations apply for the large class of isoprenylated acylphloroglucynols like 79 from H. umbraculigerum46 and 80 (linderatin) from a lauraceous Lindera species,197both isoprenylated flavonoids (chalcone and dihydrochalcone, respectively) rather than phytocannabinoids. There is little reason to consider these compounds phytocannabinoids, since their aromatic core is derived from a Claisen- and not an aldol condensation of a linear ketide, and these compounds should be better considered isoprenylated flavonoids rather than “phloroglucinyl” phytocannabinoid. The two biogenetic processes are exemplified in Scheme 13 by the structure of amorphastilbol (48g)46,173 and canniflavone 2 (= cannflavin A, 81).198 For comparison, the analogous process leading to cannabigerol is also reported. Compounds derived from both the aldol (resorcinyl) and the Claisen (phloroglucynyl) series can co-occur taxonomically unrelated plant C. sativa and H. umbraculigerum, as well as in Radula liverworts.162 Polyprenylated stilbenoids should also not “a priori” be considered “phytocannabinoids”, because this structural element is not documented within the archetypal compounds of this type from cannabis, nor should prenylated polyphenolic ketides with a hydroxylation profile different from the resorcinol one, or at least that cannot be reconduced to the further oxidation of a resorcinol core to a quinol. Compounds of this type co-occur with phytocannabinoids, e.g. demethylamorfrutin A (48a),167,169–172,174 the deoxystilbenoid 82,172 and the hydroxylated version of abnormal demethylamorfrutin A (83)178 in Radula liverworts, but the biogenetic relationship between the two groups is unclear.
 |
||
| Scheme 13 Biogenetic relationship between resorcinyl (phytocannabinoids) and phloroglucynyl (flavonoids) meroterpenoids. | ||
Finally, a compound named “dronabinol alkaloid” (84) was reported from Cassia alata L., a leguminous medicinal plant.199 The structure of this compound was only tentatively established and needs confirmation. Even if the proposed structure should be confirmed, there seems to be little reason to consider it as a cannabinoid, since plant aromatic amines are generally of anthranilate origin.
 |
||
5. Conclusions
Phytocannabinoids have a limited distribution in Nature, but occur in phylogenetically unrelated sources (higher plants, liverworts, fungi). These compounds are traditionally associated to cannabis, that, with almost 150 alkyl (C-5, C-3, C-1) phytocannabinoids reported, remains their main source of diversity. However, only a few members of the class are accumulated in substantial amounts, namely the ones having the terpenyl residue in the form of a geranyl (CBG-type), a menthyl (CBD-type and THC-type), or a prenylchromanyl (CBC-type) residue. Many of the minor cannabinoids could be auto-oxidation artifacts eventually evolving into aromatized phytocannabinoid of the CBN type, but others might be genuine natural products worth investigating from a bioactivity standpoint.
Apart from the variation of the terpenyl connectivity, structural diversity in phytocannabinoids is also related to the elongation of the isoprenyl moiety from a terpenyl- to a sesquiterpenyl moiety, while shortened analogues (hemiprenyl phytocannabinoids) have only been reported in phytocannabinoids from the aralkyl series. Oxidation of the resorcinyl moiety to a quinol is also documented, but compounds of this type have only been isolated in their acetylated and more stable form. The mammalian metabolism of phytocannabinoids involves allylic oxidation rather than nuclear oxidation to quinoid metabolites, but, due to this instability, these metabolites might have been overlooked. O-Methylation was reported in phytocannabinoids obtained from far-East samples of cannabis but it is otherwise rare in alkyl phytocannabinoids, while it is common in compounds from the phenethyl series. Aralkyl cannabinoids have a broader distribution in Nature compared to alkyl cannabinoids, but their accumulation is point-like in terms of producing organisms, with phenethyl substitution prevailing in liverworts and styryl substitution in plant constituents. Most phytocannabinoids still await an evaluation of their biological profile and pharmaceutical potential, a somewhat paradoxical observation in the light of the enormous interest for the pharmacological activity of phytocannabinoids and the messianic await for the development of cannabinoid-based medicines that permeates the media.200
It is tempting to predict that, given the biosynthetic plasticity of C. sativa, further types of alkyl phytocannabinoids will be described in the near future from both the natural and the man-induced diversity of cannabis strains. In the wake of the growing interest from amorfrutins, further additions to the phytocannabinoids inventory should also come from compounds of the aralkyl structural type. By focusing on the remarkable structural diversity of phytocannabinoids and highlighting their largely overlooked wide distribution in plants, we hope to stimulate the exploration of the biological space associated to their natural variation, going beyond the THC structural motif, and paving the way to a full opening of the Pandora’s box of their biomedical potential.
6. Acknowledgements
We would like to dedicate this article to Prof. Raphael Mechoulam, the Champollion who provided us the Rosetta stone (structure, receptors, endogenous ligands) to decipher the phytocannabinoids enigma, and a remarkable figure of a man. G. A. is grateful to Emerald Health Sciences for financial support to the laboratory.
7. References
- R. G. Pertwee, A. C. Howlett, M. E. Abood, S. P. Alexander, V. Di Marzo, M. R. Elphick, P. J. Greasley, H. S. Hansen, G. Kunos, K. Mackie, R. Mechoulam and R. A. Ross, Pharmacol. Rev., 2010, 62, 588–631 CrossRef CAS PubMed.
- R. Mechoulam and Y. Gaoni, Fortschr. Chem. Org. Naturst., 1967, 25, 175–213 CAS.
- R. Mechoulam, N. K. McCallum and S. Burstein, Chem. Rev., 1976, 76, 75–112 CrossRef CAS.
- C. E. Turner, M. A. Elsohly and E. G. Boeren, J. Nat. Prod., 1980, 43, 169–234 CrossRef CAS.
- M. A. Elsohly and D. Slade, Life Sci., 2005, 78, 539–548 CrossRef CAS PubMed.
- F. Pollastro, O. Taglialatela-Scafati, M. Allarà, E. Muñoz, V. Di Marzo, L. De Petrocellis and G. Appendino, J. Nat. Prod., 2011, 74, 2019–2022 CrossRef CAS PubMed.
- S. A. Ahmed, S. A. Ross, D. Slade, M. M. Radwan, F. Zulfiqar, R. R. Matsumoto, Y.-T. Xu, E. Viard, R. C. Speth, V. T. Karamyan and M. A. ElSohly, J. Nat. Prod., 2008, 71, 536–542 CrossRef CAS PubMed , Erratum: J. Nat. Prod. 2008, 71, 1119.
- B. Martin, Pharmacol. Rev., 1986, 38, 45–74 CAS.
- O. E. Schulz and G. Haffner, Arch. Pharm., 1960, 293, 1–6 CrossRef.
- Z. Krejči and F. Šantavý, Acta Univ. Palacki. Olomuc., Fac. Med., 1955, 6, 59–66 Search PubMed.
- C. G. Farmilo, D. T. W. McConnell, F. A. Vandenheuvel and R. Lane, United Nations Document ST/SOA/SER. S/7, 1962.
- F. Taura, S. Tanaka, C. Taguchi, T. Fukamizu, H. Tanaka, Y. Shoyama and S. Morimoto, FEBS Lett., 2009, 583, 2061–2066 CrossRef CAS PubMed.
- S. J. Gagne, J. M. Stout, E. Liu, Z. Boubakir, S. M. Clark and J. E. Page, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12811–12816 CrossRef CAS PubMed.
- X. Yang, T. Matsui, T. Kodama, T. Mori, X. Zhou, F. Taura, H. Noguchi, I. Abe and H. Morita, FEBS J., 2016, 283, 1088–1106 CrossRef CAS PubMed.
- Y. Gaoni and R. Mechoulam, Proc. Chem. Soc., 1964, 82 CAS.
- R. Mechoulam and Y. Gaoni, Fortschr. Chem. Org. Naturst., 1967, 25, 175–213 CAS.
- R. Mechoulam, Science, 1970, 168, 1159–1166 CAS.
- T. Yamauchi, Y. Shoyama, H. Aramaki, T. Azuma and I. Nishioka, Chem. Pharm. Bull., 1967, 15, 1075–1076 CrossRef CAS PubMed.
- M. Fellermeier and M. H. Zenk, FEBS Lett., 1998, 427, 283–285 CrossRef CAS PubMed.
- F. Taura, S. Sirikantaramas, Y. Shoyama, K. Yoshikai, Y. Shoyama and S. Morimoto, FEBS Lett., 2007, 581, 2929–2934 CrossRef CAS PubMed.
- F. Taura, S. Morimoto, Y. Shoyama and R. Mechoulam, J. Am. Chem. Soc., 1995, 117, 9766–9767 CrossRef CAS.
- Y. Shoyama, A. Takeuchi, F. Taura, T. Tamada, M. Adachi, R. Kuroki, Y. Shoyama and S. Morimoto, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2005, 61, 799–801 CrossRef CAS PubMed.
- F. Taura, S. Morimoto and Y. Shoyama, J. Biol. Chem., 1996, 271, 17411–17416 CrossRef CAS PubMed.
- S. Sirikantaramas, M. Morimoto, Y. Shoyama, Y. Ishikawa, Y. Wada, Y. Shoyama and F. Taura, J. Biol. Chem., 2004, 279, 39767–39774 CrossRef CAS PubMed.
- S. Morimoto, K. Komatsu, F. Taura and Y. Shoyama, J. Nat. Prod., 1997, 60, 854–857 CrossRef CAS.
- H. Itokawa, K. Takeya and S. Mihashi, Chem. Pharm. Bull., 1977, 25, 1941–1946 CrossRef CAS PubMed.
- M. Kajima and M. Piraux, Phytochemistry, 1982, 21, 67–69 CrossRef CAS.
- B. Zirpel, F. Stehle and O. Kayser, Biotechnol. Lett., 2015, 37, 1869–1875 CrossRef CAS PubMed.
- C. Onofri, E. P. M. de Meijer and G. Mandolino, Phytochemistry, 2015, 116, 57–68 CrossRef CAS PubMed.
- A. Shani and R. Mechoulam, Tetrahedron, 1974, 30, 2437–2444 CrossRef CAS.
- D. J. Harvey, Proc. Oxford Symp. Cannabis, 1985, 23–30 CAS.
- N. Raikos, H. Schmid, S. Nussbaumer, L. Ambach, S. Lanz, A. Langin, S. Konig, N. Roth, V. Auwarter and W. Weinmann, Forensic Sci. Int., 2014, 243, 130–136 CrossRef CAS PubMed.
- S. Rosenthaler, B. Pohn, C. Kolmanz, C. N. Huu, C. Krewenka, A. Huber, B. Kranner, W. D. Rausch and R. Moldzio, Neurotoxicol. Teratol., 2014, 46, 49–56 CrossRef CAS PubMed.
- R. Mechoulam and Y. Gaoni, Tetrahedron, 1965, 21, 1223–1229 CrossRef CAS PubMed.
- G. Appendino, S. Gibbons, A. Giana, A. Pagani, G. Grassi, M. Stavri, E. Smith and M. M. Rahman, J. Nat. Prod., 2008, 71, 1427–1430 CrossRef CAS PubMed.
- R. Mechoulam, Z. Ben-Zvi and Y. Gaoni, Tetrahedron, 1968, 24, 5615–5624 CrossRef CAS PubMed.
- M. M. Radwan, M. A. ElSholy, D. Slade, S. A. Ahmed, I. A. Khan and S. A. Ross, J. Nat. Prod., 2009, 72, 906–911 CrossRef CAS PubMed.
- S. Husni Afeef, R. C. McCurdy, M. M. Radwan, A. S. Ahmed, D. Slade, S. A. Ross, M. A. ElSohly and S. J. Spencer, Med. Chem. Res., 2014, 23, 4295–4300 CrossRef PubMed.
- I. Ujváry and L. Hanuš, Cannabis and Cannabinoid Research, 2016, 1, 90–101 CrossRef.
- N. M. Kogan, R. Rabinowitz, P. Levi, D. Gibson, P. Sandor, M. Schlesinger and R. Mechoulam, J. Med. Chem., 2004, 47, 3800–3806 CrossRef CAS PubMed.
- A. G. Granja, F. Carrillo-Salinas, A. Pagani, M. Gómez-Cañas, R. Negri, C. Navarrete, M. Mecha, L. Mestre, B. L. Fiebich, I. Cantarero, M. A. Calzado, M. L. Bellido, J. Fernandez-Ruiz, G. Appendino, C. Guaza and E. Muñoz, Journal of Neuroimmune Pharmacology, 2012, 7, 1002–1016 CrossRef PubMed.
- F. J. Carrillo-Salinas, C. Navarrete, M. Mecha, A. Feliú, J. A. Collado, M. L. Bellido, E. Munoz and C. Guaza, PLoS One, 2014, 9, e94733 Search PubMed.
- C. del Río, C. Navarrete, J. A. Collado, M. L. Bellido, M. Gómez-Cañas, M. R. Pazos, J. Fernández-Ruiz, F. Pollastro, G. Appendino, M. A. Calzado, I. Cantarero and E. Muñoz, Sci. Rep., 2016, 6, 29789, DOI:10.1038/srep21703.
- L. W. Robertson, M. A. Lyle and S. Billets, Biomed. Mass Spectrom., 1975, 2, 266–271 CrossRef CAS.
- E. Stern and D. M. Lambert, Chem. Biodiversity, 2007, 4, 1707–1728 CAS.
- F. Bohlmann and E. Hoffmann, Phytochemistry, 1979, 18, 1371–1374 CrossRef CAS.
- L. De Petrocellis, V. Vellani, A. Schiano-Moriello, P. Marini, P. C. Magherini, P. Orlando and V. Di Marzo, J. Pharmacol. Exp. Ther., 2008, 325, 1007–1015 CrossRef CAS PubMed.
- M. G. Cascio, L. A. Gauson, L. A. Stevenson, R. A. Ross and R. G. Pertwee, Br. J. Pharmacol., 2010, 159, 129–141 CrossRef CAS PubMed.
- Z. P. Khan, C. N. Ferguson and R. M. Jones, Anaesthesia, 1999, 54, 146–165 CrossRef CAS PubMed.
- Y. Tatsuo, S. Yukihiro, M. Yoko and N. Itsuo, Chem. Pharm. Bull., 1968, 16, 1164–1165 CrossRef.
- G. Appendino, A. Giana, S. Gibbons, M. Maffei, G. Gnavi, G. Grassi and O. Sterner, Nat. Prod. Commun., 2008, 3, 1977–1980 CAS.
- M. M. Radwan, S. A. Ross, D. Slade, S. A. Ahnmed, F. Zulfiqar and M. A. ElSohly, Planta Med., 2008, 74, 267–272 CrossRef CAS PubMed.
- M. M. Radwan, M. A. ElSohly, D. Slade, S. A. Ahmed, L. Wilson, A. T. El-Alfy, I. A. Khan and S. A. Ross, Phytochemistry, 2008, 69, 2627–2633 CrossRef CAS PubMed.
- C. Weidner, J. C. de Groot, A. Prasad, A. Freiwald, C. Quedenau, M. Kliem, V. Kodelja, C. T. Han, S. Giegold, M. Baumann, B. Klebl, K. Siems, L. Müller-Kuhrt, A. Schürmann, R. Schüler, A. F. Pfeiffer, F. C. Schroeder, K. Büssow and S. Sauer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7257–7262 CrossRef CAS PubMed.
- M. Srebnik, N. Lander and R. Mechoulam, J. Chem. Soc., Perkin Trans. 1, 1984, 2881–2886 RSC.
- S. Yukihiro, H. Hitotoshi, O. Miyuki, S. Takao and N. Itsuo, Chem. Pharm. Bull., 1975, 23, 1894–1895 CrossRef.
- Y. Shoyama, H. Hirano, H. Makino, N. Umekita and I. Nishioka, Chem. Pharm. Bull., 1977, 25, 2306–2311 CrossRef CAS.
- Y. Shoyama, T. Yamauchi and I. Nishioka, Chem. Pharm. Bull., 1970, 18, 1327–1332 CrossRef CAS.
- F. Taura, S. Morimoto and Y. Shoyama, Phytochemistry, 1995, 39, 457–458 CrossRef CAS.
- L. O. Hanuš, R. Levy, D. De La Vegga, L. Katz, M. Roman and P. Tomíček, Isr. J. Plant Sci., 2016, 63, 182–190 Search PubMed.
- A. S. Husni, C. R. McCurdy, M. M. Radwan, A. S. Ahmed, D. Slade, S. A. Ross, M. A. ElSohly and J. S. Cutler, Med. Chem. Res., 2014, 23, 4295–4300 CrossRef CAS PubMed.
- Y. Gaoni and R. Mechoulam, Chem. Commun., 1966, 20–21 RSC.
- U. Claussen, F. v. Spulak and F. Korte, Tetrahedron, 1966, 22, 1477–1479 CrossRef CAS.
- A. A. Izzo, F. Borrelli, R. Capasso, V. Di Marzo and R. Mechoulam, Trends Pharmacol. Sci., 2009, 30, 515–527 CrossRef CAS PubMed.
- R. K. Razdan, in The Total Synthesis of Natural Products, ed. J. ApSimon, 2007, vol. 4, pp. 185–262 Search PubMed.
- K. Quaghebeur, J. Coosemans, S. Toppet and F. Compernolle, Phytochemistry, 1994, 37, 159–161 CrossRef CAS PubMed.
- (a) N. Iwata, N. Wang, X. Yao and S. J. Kitanaka, J. Nat. Prod., 2004, 67, 1106–1109 CrossRef CAS PubMed; (b) N. Iwata and S. Kitanaka, Chem. Pharm. Bull., 2011, 59, 1409–1412 CrossRef CAS PubMed; (c) V. Hellwig, R. Nopper, F. Mauler, J.-K. Liu, Z.-H. Ding and M. Stadler, Arch. Pharm. (Weinheim, Ger.), 2003, 336, 119–126 CrossRef CAS PubMed.
- R. A. De Zeewen, T. B. Vree, D. D. Breimer and C. A. M. Van Ginneken, Experientia, 1973, 29, 260–261 CrossRef.
- Y. Shoyama, F. Toshio, Y. Tatsuo and N. Itsuo, Chem. Pharm. Bull., 1968, 24, 7–12 Search PubMed.
- (a) R. Adams, B. R. Baker and R. B. Wearn, J. Am. Chem. Soc., 1940, 62, 2204–2207 CrossRef CAS; (b) A. Jacob and A. Todd, Nature, 1940, 350 CrossRef CAS.
- R. Mechoulam and Y. Shvo, Tetrahedron, 1963, 19, 2073–2078 CrossRef CAS.
- F. Šantavý, Acta Univ. Palacki. Olomuc., Fac. Med., 1964, 35, 5–9 Search PubMed.
- (a) T. Petrzilka, W. Haeffliger, C. Sikemeier, G. Ohloff and A. Eschenmoser, Helv. Chim. Acta, 1967, 50, 719–723 CrossRef CAS PubMed; (b) T. Petrzilka, W. Haeffliger and C. Sikemeier, Helv. Chim. Acta, 1969, 52, 1102–1134 CrossRef CAS.
- P. Seephonkai, R. Papescu, M. Zehl, G. Krupitza, E. Urban and B. Kopp, J. Nat. Prod., 2011, 74, 712–717 CrossRef CAS PubMed.
- (a) P. G. Jones, L. Falvello, O. Kennard and R. Mechoulam, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 3211–3214 CrossRef; (b) T. Ottersen, E. Rosenqvist, C. E. Turner and F. S. El-Feraly, Acta Chem. Scand., Ser. B, 1977, 31, 807–812 CrossRef CAS. For a detailed analysis of atropisomerism in CBD, see: (c) I. Tamir and R. Mechoulam, J. Med. Chem., 1980, 23, 220–223 CrossRef CAS PubMed; (d) V. V. Kane, A. R. Martin, C. Jaime and E. Osawa, Tetrahedron, 1984, 41, 2919–2927 CrossRef; (e) H. Berber, P. Lameiras, C. Denhez, C. Antheume and J. Clayden, J. Org. Chem., 2014, 79, 6015–6027 CrossRef CAS PubMed.
- J. Merrick, B. Lane, T. Sebree, T. Yaksj, C. O’Neill and S. L. Banks, Cannabis and Cannabinoid Research, 2016, 1, 102–112 CrossRef.
- S. Agurell and K. Leander, Acta Pharm. Suec., 1971, 8, 391–402 CAS.
- F. J. E. M. Küppers, R. J. J. C. Sousberg, C. A. L. Bercht, C. A. Salemink, J. K. Terlouw, W. Heerma and A. Laven, Tetrahedron, 1973, 29, 2797–2802 CrossRef.
- F. von Spulak, U. Claussen and H.-W. Fehlhaber, Tetrahedron, 1968, 24, 5379–5383 CrossRef CAS PubMed.
- R. B. Laprairie, A. M. Bagher, M. E. Kelly and E. M. Denovan-Wright, Br. J. Pharmacol., 2015, 172, 4790–4805 CrossRef CAS PubMed.
- W. B. O’Shaughnessy, Transactions of the Medical and Physical Society of Bengal, 1838-1840, vol. 8, pp. 462–469 Search PubMed.
- T. B. Vree, D. D. Breimer, C. A. M. Van Ginneken and J. M. Van Rossum, J. Pharm. Pharmacol., 1972, 24, 7–12 CrossRef CAS PubMed.
- L. Vollner, D. Bienik and F. Korte, Tetrahedron Lett., 1969, 10, 145–147 CrossRef.
- D. J. Harvey, J. Pharm. Pharmacol., 1976, 28, 280–285 CrossRef CAS PubMed.
- R. M. Smith, J. Forensic Sci., 1997, 42, 610–618 CAS.
- Y. Shoyama, K. Kuboe, I. Nishioka and T. Yamauchi, Chem. Pharm. Bull., 1972, 20, 2072 CrossRef CAS.
- H. Hendriks, T. M. Malingre, S. Batterman and R. Bos, Pharm. Weekbl., 1978, 113, 413–424 CAS.
- C. A. M. van Ginneken, T. B. Vree, D. D. Breimer, H. W. H. Thijssen and J. M. van Rossum, Proc. Int. Symp. on GC-MS, Elba, Italy, 1972, p. 109 Search PubMed.
- J. Friedrich-Fiechtl and G. Spiteller, Tetrahedron, 1975, 31, 479–487 CrossRef CAS.
- A. Pagani, F. Scala, G. Chianese, G. Grassi, G. Appendino and O. Taglialatela-Scafati, Tetrahedron, 2011, 67, 3369–3373 CrossRef CAS.
- T. B. Vree, D. D. Breimer, C. A. M. Van Ginneken and J. M. Van Rossum, J. Chromatogr., 1972, 74, 209–224 CrossRef CAS PubMed.
- R. J. J. C. Lousberg, C. A. L. Bercht, R. Vannoyen and H. J. W. Spronck, Phytochemistry, 1977, 16, 595–597 CrossRef CAS.
- E. C. Taylor, K. Lenard and Y. Shvo, J. Am. Chem. Soc., 1966, 88, 367–369 CrossRef CAS.
- T. Petrzilka and C. Sikemeier, Helv. Chim. Acta, 1967, 50, 2011–2013 CrossRef.
- R. Mechoulam, P. Braun and Y. Gaoni, J. Am. Chem. Soc., 1967, 89, 4552–4554 CrossRef CAS PubMed.
- J. Zias, H. Stark, J. Seligman, R. Levy, E. Werker, A. Breuer and R. Mechoulam, Nature, 1993, 363, 215 CAS.
- R. L. Hiverly, W. A. Mosher and F. W. Hoffman, J. Am. Chem. Soc., 1966, 88, 1832–1833 CrossRef.
- L. Hanuš and Z. Krejčí, Acta Univ. Palacki. Olomuc., Fac. Med., 1975, 74, 161–166 Search PubMed.
- S. A. Ross, M. M. Radwan, A. T. El-Alfy, S. P. Manly, S. A. Ahmed, D. Slade, A. Eslinger, L. Wilson, S. Seale, O. Dale, S. Cutler, I. A. Khan and M. M. ElSohly, 20thAnnual Symposium of the International Cannabinoid Research Society, Research Triangle Park, NC, USA, 2010, pp. P4–21 Search PubMed.
- S. A. Ross, D. Slade, S. A. Ahmed, M. M. Radwan and M. A. ElSohly, 18th Annual Symposium of the International Cannabinoid Research Society, Aviemore, 2008, p. P141 Search PubMed.
- H. J. Wollner, J. R. Matchett, J. Levine and S. Loewe, J. Am. Chem. Soc., 1942, 64, 26–29 CrossRef CAS.
- Y. Gaoni and R. Mechoulam, J. Am. Chem. Soc., 1964, 86, 1646–1647 CrossRef CAS.
- L. Tudge, C. Williams, P. J. Cowen and C. McCabe, Int. J. Neuropsychopharmacol., 2014, 18 DOI:10.1093/ijnp/pyu094.
- D. B. Uliss, H. C. Dalzell, G. R. Handrick, J. F. Howes and R. K. Razdan, J. Med. Chem., 1975, 18, 213–215 CrossRef CAS PubMed.
- C. E. Turner, K. W. Hadley, B. F. J. Henry and M. L. Mole Jr, J. Pharm. Sci., 1974, 63, 1872–1876 CrossRef CAS PubMed.
- C. E. Turner, K. W. Hadley, P. S. Fetterman, N. J. Doorenbos, M. W. Quimby and C. Waller, J. Pharm. Sci., 1973, 62, 1601–1605 CrossRef CAS PubMed.
- I. J. Miller, N. K. McCallum, C. M. Kirk and B. M. Peake, Experientia, 1982, 38, 230–231 CrossRef CAS.
- S. A. Ross, M. A. El-Sohly, G. N. N. Sultana, Z. Mehmedic, C. F. Hossain and S. Chandra, Phytochem. Anal., 2005, 16, 45–48 CrossRef CAS PubMed.
- H. G. Pars and R. K. Razdan, Ann. N. Y. Acad. Sci., 1971, 191, 15–22 CrossRef CAS.
- E. W. Gill, J. Chem. Soc. C, 1971, 579–582 RSC.
- C. E. Turner, K. Hadley and P. S. Fetterman, J. Pharm. Sci., 1973, 62, 1739–1741 CrossRef CAS PubMed.
- F. Korte, M. Haag and U. Claussen, Angew. Chem., Int. Ed., 1965, 4, 872 CrossRef CAS.
- R. Mechoulam, Z. Ben-Zvi, B. Yagnitinsky and A. Shani, Tetrahedron Lett., 1969, 2339–2343 CrossRef CAS.
- M. N. Qureshi, F. Kanwal, M. Siddique, I.-u. Rahman and M. Akram, World Appl. Sci. J., 2012, 19, 918–923 CAS.
- E. G. Boeren, M. A. ElSohly and C. E. Turner, Experientia, 1979, 35, 1278–1279 CrossRef CAS PubMed.
- H. N. ElSohly, E. G. Boeren, C. E. Turner and M. A. ElSohly, in Cannabinoids: Chemistry, Pharmacology and Therapeutic Aspects, ed. S. Agurell, W. L. Dewey and R. E. Willette, 1984, pp. 89–96 Search PubMed.
- F. Zulfiqar, S. A. Ross, D. Slade, S. A. Ahmed, M. M. Radwan, Z. Ali and I. A. Khan, Tetrahedron Lett., 2012, 53, 3560–3562 CrossRef CAS PubMed.
- R. M. Smith and K. D. Kempfert, Phytochemistry, 1977, 16, 1088–1089 CrossRef CAS.
- D. B. Uliss, G. R. Handrick, H. C. Dalzell and R. K. Razdan, Tetrahedron, 1978, 34, 1885–1888 CrossRef CAS.
- A. V. Malkov and P. Kocovsky, Collect. Czech. Chem. Commun., 2001, 66, 1257–1268 CrossRef CAS.
- M. A. Schafroth, G. Zuccarello, S. Krautwald, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2014, 53, 13898–13901 CrossRef CAS PubMed.
- L. Crombie and R. Ponsford, J. Chem. Soc. C, 1971, 796–784 RSC.
- C. A. L. Bercht, R. J. J. C. Lousberg, J. E. M. Küppers and C. A. Salemink, Phytochemistry, 1974, 13, 619–621 CrossRef CAS.
- R. Adams, Harvey Lect., 1942, 37, 168–197 Search PubMed , also appeared as R. Adams, Bull. N. Y. Acad. Med. 1942, 18, 705-730.
- A. R. Todd, Experientia, 1946, 2, 55–60 CrossRef CAS PubMed.
- L. E. Hollister, H. K. Gillespie, R. Mechoulam and M. Srebnik, Psychopharmacology, 1987, 92, 505–507 CrossRef CAS PubMed.
- M. A. ElSohly, E. G. Boeren and C. E. Turner, Experientia, 1978, 34, 1127–1128 CrossRef CAS PubMed.
- Y. Obata and Y. Ishikawa, Agric. Biol. Chem., 1966, 30, 619–620 CAS.
- W. R. Chan, K. E. Magnus and H. A. Watson, Experientia, 1976, 32, 283–284 CrossRef CAS PubMed.
- M. A. ElSohly, F. S. El-Feraly and C. E. Turner, Lloydia, 1977, 40, 275–278 CAS.
- Y. Shoyama, S. Morimoto and I. Nishioka, Chem. Pharm. Bull., 1981, 29, 3720–3723 CrossRef CAS.
- M. Mariko and A. Hiroaki, Kagaku Keisatsu Kenkyusho Hokoku, Hokagaku-hen, 1984, 37, 137–140 Search PubMed.
- C. E. Turner, M. L. Mole, L. Hanuš and H. N. El-Sohly, J. Nat. Prod., 1981, 44, 27–33 CrossRef CAS.
- F. Korte and H. Sieper, J. Chromatogr. A, 1964, 13, 90–98 CrossRef CAS PubMed.
- D. A. Whiting, M. J. Bengley, D. G. Clarke and L. Crombie, J. Chem. Soc., Chem. Commun., 1970, 1547–1548 CAS.
- Y. Kashiwada, K. Yamazaki, Y. Ikeshiro, T. Yamagishi, T. Fujioka, K. Mihashi, K. Mizuki, L. M. Cosentino, K. Fowke, S. L. Morris-Natschke and K. H. Lee, Tetrahedron, 2011, 57, 1559–1563 CrossRef.
- T. B. Vree, D. D. Breimer, C. A. M. van Ginneken and J. N. van Rossum, J. Chromatogr., 1972, 74, 124–127 CrossRef CAS PubMed.
- L. Crombie and R. Ponsford, Chem. Commun., 1968, 894–895 RSC.
- Y. Shoyama, O. Reiko, Y. Tatsuo and N. Itsuo, Chem. Pharm. Bull., 1972, 20, 1927–1930 CrossRef CAS.
- A. Shani and R. Mechoulam, Chem. Commun., 1970, 273–274 RSC.
- D. B. Uliss, R. K. Razdan and H. C. Dalzell, J. Am. Chem. Soc., 1974, 96, 7372–7374 CrossRef CAS PubMed.
- I. Yamamoto, H. Gohda, S. Narimatsu, K. Watanabe and H. Yoshimura, Pharmacol., Biochem. Behav., 1991, 40, 541–546 CrossRef CAS.
- S. C. Hartsel, W. H. Loh and L. W. Robertson, Planta Med., 1983, 48, 17–19 CrossRef CAS PubMed.
- C. A. L. Bercht, R. J. J. Lousberg, F. J. E. M. Kuppers, C. A. Salemink and T. B. Vree, J. Chromatogr., 1973, 81, 163–166 CrossRef CAS PubMed.
- T. B. Wood, W. T. N. Spivey and T. H. Easterfield, J. Chem. Soc., 1896, 69, 539–546 RSC.
- E. B. Russo, H.-E. Jiang, X. Li, A. Sutton, A. Carboni, F. del Bianco, G. Mandolino, D. J. Potter, Y.-X. Zhao, S. Bera, Y.-B. Zhang, E.-G. Lü, D. K. Ferguson, F. Hueberl, L.-C. Zhao, C.-J. Liu, Y.-F. Wang and C. S. J. Li, Exp. Bot., 2008, 59, 4171–4182 CrossRef CAS PubMed.
- R. Adams, M. Hunt and J. H. Clark, J. Am. Chem. Soc., 1940, 62, 196–200 CrossRef CAS.
- B. K Prasad, A. Hazekamp and R. Verpoorte, Planta Med., 2007, 73, 273–275 CrossRef PubMed.
- F. W. Merkus, Nature, 1971, 232, 579–580 CrossRef CAS PubMed.
- H. Grote and G. Spiteller, J. Chromatogr., 1978, 154, 13–23 CrossRef CAS.
- H. Grote and G. Spiteller, Tetrahedron, 1978, 34, 3207–3213 CrossRef CAS.
- S. A. Ahmed, S. A. Ross, D. Slade, M. M. Radwan, I. A. Khan and M. A. ElSohly, Tetrahedron Lett., 2008, 49, 6050–6053 CrossRef CAS PubMed.
- O. Taglialatela-Scafati, A. Pagani, F. Scala, L. De Petrocellis, V. Di Marzo, G. Grassi and G. Appendino, Eur. J. Org. Chem., 2010, 11, 2067–2072 CrossRef.
- J. Carreras, M. S. Kirillova and A. M. Echavarren, Angew. Chem., Int. Ed., 2016, 55, 7121–7125 CrossRef CAS PubMed.
- S. Sauer, ChemBioChem, 2014, 15, 1231–1238 CrossRef CAS PubMed.
- L. Fuhr, M. Roussearu, A. Plauth, F. C. Schroeder and S. Sauer, J. Nat. Prod., 2015, 78, 1160–1164 CrossRef CAS PubMed.
- C. Weidner, J. C. de Groot, A. Prasad, A. Freiwald, C. Quedenau, M. Kliem, A. Witzke, V. Kodelja, C. T. Han, S. Giegold, M. Baumann, B. Klebl, K. Siems, L. Müller-Kuhrt, A. Schürmann, R. Schüler, A. F. Pfeiffer, F. C. Schroeder, K. Büssow and S. Sauer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7257–7262 CrossRef CAS PubMed.
- Y. Asakawa, M. Toyota and T. Takemoto, Phytochemistry, 1978, 17, 2005–2010 CrossRef CAS.
- L. A. Mitscher, Y. H. Park, A. Al Shamma, P. B. Hudson and T. Haas, Phytochemistry, 1981, 20, 781–785 CrossRef CAS.
- E. L. Ghisalberti, P. R. Jefferies and D. McAdam, Phytochemistry, 1981, 20, 1959–1961 CrossRef CAS.
- K. P. Manfredi, V. Vallurupalli, M. Demidova, K. Kindscher and L. K. Pannell, Phytochemistry, 2001, 58, 153–157 CrossRef CAS PubMed.
- Y. Asakawa, Prog. Chem. Org. Nat. Prod., 1995, 66, 1–562 Search PubMed.
- C. Chen, Y. Wu and L. Du, Pharm. Biol., 2016, 54, 488–493 CrossRef CAS PubMed.
- J. C. de Groot, C. Weidner, J. Krausze, K. Kawamoto, F. C. Schroeder, S. Sauer and K. Büssow, J. Med. Chem., 2013, 56, 1535–1543 CrossRef CAS PubMed.
- H. W. Choi, M. Tian, M. Manohar, M. M. Harraz, S. Park, F. C. Schroeder, S. H. Snyder and D. Klessig, PLoS One, 2015, 10, e0143447 Search PubMed.
- M. Kemal, S. K. Wahba Khalil, N. G. Rao and N. F. Woolsey, J. Nat. Prod., 1979, 42, 463–468 CrossRef CAS.
- W. Lee, J. Ham, H. C. Kwon, Y. K. Kim and S. N. Kim, Biochem. Biophys. Res. Commun., 2013, 432, 73–79 CrossRef CAS PubMed.
- V. S. Sobolev, N. M. Krausert and J. B. J. Gloer, J. Agric. Food Chem., 2016, 64, 579–584 CrossRef CAS PubMed.
- Y. Asakawa, E. Kusube, T. Takemoto and C. Sure, Phytochemistry, 1979, 18, 1371–1373 CrossRef.
- L. A. Mitscher, G. S. Raghav Rao, I. Khanna, T. Veysoglu and S. Drake, Phytochemistry, 1983, 22, 573–576 CrossRef CAS.
- Y. Asakawa, E. Kusube, T. Takemoto and C. Sure, Phytochemistry, 1978, 17, 2115–2117 CrossRef CAS.
- Y. Asakawa, T. Hashimoto, K. T. Takikawa, M. Tori and S. Ogawa, Phytochemistry, 1991, 30, 235–251 CrossRef CAS.
- L. A. Mitscher, S. R. Gollapudi, S. Drake and D. S. Oburn, Phytochemistry, 1985, 24, 1481–1483 CrossRef CAS.
- F. Cullmann and H. Becker, Z. Naturforsch., C: J. Biosci., 1999, 54, 147–150 CAS.
- Y. Asakawa, K. Kondo and M. Tori, Phytochemistry, 1991, 30, 325–328 CrossRef CAS.
- J. R. Joset, A. Marston, M. P. Gupta and K. Hostettmann, J. Nat. Prod., 2001, 64, 710–715 CrossRef.
- M. Toyota, T. Shimamura, H. Ishi, M. Renner, J. Braggins and Y. Asakawa, Chem. Pharm. Bull., 2002, 50, 1390–1392 CrossRef CAS PubMed.
- Y. Asakawa, K. Kondo, M. Tori, T. Hashimoto and S. Ogawa, Phytochemistry, 1991, 30, 219–234 CrossRef CAS.
- L. Kraut, R. Mues and H. D. Zinsmeister, Phytochemistry, 1997, 45, 1249–1255 CrossRef CAS.
- F. Nagashima and Y. Asakawa, Molecules, 2011, 16, 10471–10478 CrossRef PubMed.
- M. Toyota, T. Kinugawa and Y. Asakawa, Phytochemistry, 1994, 37, 859–862 CrossRef CAS.
- L. Harinantenaina, Y. Takahara, T. Nishizawa, C. Kochi, G. L. Soma and Y. Asakawa, Chem. Pharm. Bull., 2006, 54, 1046–1049 CrossRef CAS PubMed.
- A. Abu-Mellal, N. Koolaji, R. K. Duke, V. H. Tran and C. C. Duke, Phytochemistry, 2012, 77, 251–259 CrossRef CAS PubMed.
- C. M. Starks, R. B. Williams, V. L. Norman, S. M. Rice, M. O’Neil-Johnson, J. A. Lawrence and G. R. Eldridge, Phytochemistry, 2014, 98, 216–222 CrossRef CAS PubMed.
- I. Muhammaed, X.-C. Li, M. L. Jacob, B. L. Tekawani, D. C. Dumbar and D. Ferreira, J. Nat. Prod., 2003, 66, 804–809 CrossRef PubMed.
- I. Muhammad, X.-C. Li, D. C. Dunbar, M. A. ElSohly and I. A. Khan, J. Nat. Prod., 2001, 64, 1322–1325 CrossRef CAS.
- L. W. Crombie, Pure Appl. Chem., 1986, 58, 693–700 CrossRef CAS.
- L. W. Crombie, M. L. Crombie and D. F. Firth, J. Chem. Soc., Perkin Trans. 1, 1988, 1263–1270 RSC.
- Y. Song, S. Hwang, P. Gong, D. Kim and S. Kim, Org. Lett., 2008, 10, 269–271 CrossRef CAS PubMed.
- See, for instance: http://www.bluelight.org/vb/archive/index.php/t-642032.html, accessed on March 7, 2016.
- Q. Wang, Q. Huang, B. Chen, J. Lu, H. Wang, X. She and X. Pan, Angew. Chem., Int. Ed., 2006, 45, 3651–3653 CrossRef CAS PubMed.
- D. H. Dehte, R. D. Erande, S. Mahapatra, S. Das and B. Kumar, Chem. Commun., 2015, 51, 2871–2873 RSC.
- F. Klotter and A. Studer, Angew. Chem., Int. Ed., 2015, 54, 1–5 CrossRef.
- G. Delle Monache, B. Botta, V. Vinciguerra, J. F. De Mello and A. De Andrade Chiappeta, Phytochemistry, 1996, 41, 537–544 CrossRef CAS.
- B. Botta, E. Gacs-Baitz, V. Vinciguerra and G. Delle Monache, Phytochemistry, 2003, 64, 599–602 CrossRef CAS PubMed.
- T. Tanaka, M. Ohyama, Y. Kawasaka and M. Iinuma, Tetrahedron Lett., 1994, 35, 9043–9044 CrossRef CAS.
- H. Tanaka, K. Ichino and K. Ito, Chem. Pharm. Bull., 1984, 32, 3747–3750 CrossRef CAS.
- (a) L. Crombie, W. M. Crombie and S. V. Jamieson, Tetrahedron Lett., 1980, 21, 3607–3610 CrossRef CAS; (b) B. Barrett, A. M. Scutt and F. J. Evans, Experientia, 1986, 42, 452–453 CrossRef PubMed.
- D. E. Okwu and F. U. Nnamdi, Chem. Sin., 2011, 2, 247–254 CAS.
- E. B. Russo, Trends Pharmacol. Sci., 2016, 37, 594–560 CrossRef CAS PubMed.
|
|
| This journal is © The Royal Society of Chemistry 2016 |

