

Visual Overview

Abstract
 The metabotropic glutamate 5 receptor and the cannabinoid type 1 receptor are G protein–coupled receptors that are widely expressed in the central nervous system. Metabotropic glutamate 5 receptors, present at the postsynaptic site, are coupled to Gαq/11 proteins and display an excitatory response upon activation, whereas the cannabinoid type 1 receptor, mainly present at presynaptic terminals, is coupled to the Gi/o protein and triggers an inhibitory response. Recent studies suggest that the glutamatergic and endocannabinoid systems exhibit a functional interaction to modulate several neural processes. In this review, we discuss possible mechanisms involved in this crosstalk and its relationship with physiologic and pathologic conditions, including nociception, addiction, and fragile X syndrome.
The metabotropic glutamate 5 receptor and the cannabinoid type 1 receptor are G protein–coupled receptors that are widely expressed in the central nervous system. Metabotropic glutamate 5 receptors, present at the postsynaptic site, are coupled to Gαq/11 proteins and display an excitatory response upon activation, whereas the cannabinoid type 1 receptor, mainly present at presynaptic terminals, is coupled to the Gi/o protein and triggers an inhibitory response. Recent studies suggest that the glutamatergic and endocannabinoid systems exhibit a functional interaction to modulate several neural processes. In this review, we discuss possible mechanisms involved in this crosstalk and its relationship with physiologic and pathologic conditions, including nociception, addiction, and fragile X syndrome.
Introduction
Glutamate, the most important excitatory neurotransmitter in the central nervous system (CNS) of mammals, modulates several neural events, including development and cognition, as well as neurologic pathogenesis. Glutamate exerts its actions by binding and activating glutamatergic receptors, which are classified into two distinct groups: ionotropic glutamatergic receptors and metabotropic glutamatergic receptors (mGlu receptors). The ionotropic glutamatergic receptors are ligand-gated ion channels and are subdivided into three types: N-methyl-d-aspartate receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors, and kainate receptors (Karakas et al., 2015). mGlu receptors are G protein–coupled receptors that can trigger cell signaling transduction pathways through intracellular second messengers to modulate synaptic activity. Eight different mGlu receptors are subdivided into three groups (I, II, and III) based on signal transduction, sequence homology, and pharmacological properties (Nakanishi, 1992). Group I includes the mGlu1and mGlu5 receptors, which mediate excitatory responses through the activation of phosphoinositide signaling pathways, and group II and III receptors, which inhibit the cAMP signaling pathway (Nakanishi, 1992).
mGlu5 Receptor: Pharmacology and Cell Signaling Pathways
Several studies indicate that the mGlu5 receptor is central to various brain processes, including long-term potentiation and long-term depression (LTD), mechanisms important for memory acquisition and learning (Bliss and Collingridge, 1993; Sung et al., 2001; Hou and Klann, 2004; Hullinger et al., 2015). Moreover, relevant neuronal processes such as cell differentiation, plastic alterations, and cell death are also modulated by mGlu receptors (Sheng and Kim, 2002; Erichsen et al., 2015). The mGlu5 receptor exhibits a three-dimensional structure commonly observed among mGlu receptor family members, including a large extracellular amino-terminal portion where the orthosteric site is located, a transmembrane heptahelical domain that contains an allosteric binding site, and an intracellular C-terminal portion (Nakanishi and Masu, 1994). The mGlu5 receptor is broadly expressed in the CNS and is predominantly located perisynaptically near postsynaptic elements and in glial cells (Shigemoto et al., 1993; Nusser et al., 1994; Lujan et al., 1996; Cai et al., 2000). Notably, the mGlu5 receptor is expressed in different brain regions, including the olfactory bulb, anterior olfactory nuclei, olfactory tubercle, cerebral cortex, hippocampus, septum side, striatum, nucleus accumbens, inferior colliculus, and spinal trigeminal nuclei (Abe et al., 1992).
mGlu5 receptors can activate multiple cell signaling pathways (Fig. 1). The classic canonical form of the mGlu5 receptor signaling starts with glutamate binding to the orthosteric site of the receptor. The ligand/receptor interaction induces Gαq/11 protein activation, which leads to the activation of phospholipase C (PLC)-β and the formation of second messengers, including diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). IP3 binds to its receptor in the endoplasmic reticulum (IP3R), and this leads to an increase in intracellular Ca2+ levels and consequent activation of protein kinase C (Berridge and Irvine, 1984). Simultaneously, protein kinase C activation drives mGlu5 receptor desensitization through phosphorylation (Alaluf et al., 1995; Dhami and Ferguson, 2006; Ribeiro et al., 2009, 2010).

Cell signaling pathways activated by the mGlu5 receptor. The scheme illustrates an overview of mGlu5 receptor cell signaling pathways. After glutamate binding, the mGlu5 receptor is stimulated and promotes G-protein dissociation, which activates PLCβ and leads to DAG cleavage and formation of the second messengers IP3 and DAG. IP3 binds to and stimulates IP3Rs, increasing intracellular Ca2+ levels and contributing to ERK1/2 activation. In addition, the C-terminal portion of the mGlu5 receptor can interact with Homer proteins that interact with other protein partners to trigger various biologic cascades such as formation of the endocannabinoid 2-AG and phosphorylation of AKT and ERK1/2. Moreover, activation of these distinct pathways underlies important biologic processes, including cell proliferation and survival. Protein–protein interactions are represented by arrow-chains and activated elements are represented by continuous arrows. Glu, glutamate; MEK, mitogen-activated protein kinase kinase; PIKE, phosphoinositide 3-kinase enhancer.
mGlu5 receptor activation can facilitate its interaction with postsynaptic proteins, triggering the activation of other cell signaling cascades (Tu et al., 1999). Homer-type proteins are mainly postsynaptic, acting as scaffold proteins and binding directly to the C-terminal portion of the mGlu5 receptor (Brakeman et al., 1997). Notably, Homer proteins appear to be involved in the anchorage and distribution of mGlu 5 receptor at the periphery of the synapse and in the expression of mGlu5 receptors at the plasma membrane (Roche et al., 1999; Ango et al., 2002; Mansouri et al., 2015). Moreover, mGlu5/Homer interaction may also interfere with the regulation of synaptic transmission, because previous studies show that NMDAR activity is controlled by the mGlu5 receptor through the protein complex formed by Homer (Moutin et al., 2012). Moreover, Homer proteins may control spontaneous activity of group I mGlu receptors (Ango et al., 2001). Homer proteins are widely expressed in neurons and form complexes with postsynaptic density proteins to facilitate the interaction between plasma membrane receptors and intracellular signal transduction components (Xiao et al., 1998; Shiraishi-Yamaguchi and Furuichi, 2007). For example, Homer binding to IP3Rs brings the mGlu5 receptor into close proximity, facilitating the activation of IP3Rs and the release of Ca2+ from intracellular stores (Tu et al., 1998). mGlu5 receptor activation can trigger phosphorylation of extracellular signal-regulated kinases (ERK1/2) and part of this effect is attributable to signaling mediated by IP3/Ca2+ (Mao et al., 2005; Wang et al., 2007; Jong et al., 2009). However, it has been shown that Homer establishes an alternative signaling pathway linking the mGlu5 receptor to ERK1/2 in a Ca2+-independent manner (Fig. 1) (Mao et al., 2005). Moreover, both IP3/Ca2+- and Homer-dependent cascades are necessary to activate ERK1/2 to levels high enough to phosphorylate the transcription factors ETS domain-containing protein-1 and cAMP response element-binding protein, facilitating the expression of genes involved in cell proliferation (Mao et al., 2005). It has also been shown that activation of the mGlu5 receptor in mouse hippocampal slices results in phosphoinositide 3-kinase (PI3K)–dependent protein kinase B (AKT) phosphorylation (Hou and Klann, 2004; Ribeiro et al., 2010). The mechanism underlying mGlu5-dependent AKT activation has been elucidated, and it has been reported that activation of mGlu receptors enhances formation of the complex mGlu5 receptor, Homer, and PI3K enhancer, which leads to activation of PI3K (Rong et al., 2003). PI3K increases the levels of phosphatidylinositol 3,4,5-trisphosphate, facilitating the recruitment of pyruvate dehydrogenase kinase 1 (PDK1) and AKT to the cell membrane, where AKT is phosphorylated by pyruvate dehydrogenase kinase 1 (Burgering and Coffer, 1995; Rong et al., 2003). AKT activation triggers important downstream signaling events, such as apoptosis inhibition and transcription activation of genes involved in cell proliferation and survival (Song et al., 2005).
Various synthetic ligands were found to interact with the mGlu5 receptor, facilitating the understanding of many different downstream signaling effects triggered by this receptor. 3,5-Dihydroxyphenylglycine (DHPG) is an orthosteric ligand that can activate both mGlu5 and mGlu1 receptors (Ito et al., 1992; Fitzjohn et al., 1996). Further studies led to the discovery of (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), which was at first proposed as a mGlu5 receptor–selective agonist but was shown to also activate mGlu1receptors (Doherty et al., 1997; Kammermeier, 2012). Another class of compounds that act as positive allosteric modulators can bind to an allosteric regulatory site that is topographically distinct from the conserved orthosteric agonist binding site, and they can thus be more selective for the different mGlu receptor subtypes since the allosteric site is weakly conserved (Sheffler et al., 2011). Interestingly, the mGlu5 receptor positive allosteric modulator, 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB), can activate neuroprotective cell signaling pathways without increasing intracellular Ca2+(Doria et al., 2013). Moreover, CDPPB can rescue neuronal cell death, abrogate huntingtin aggregation, and prevent memory deficit in a mouse model of Huntington disease (Doria et al., 2015). The widely used mGlu5receptor inhibitor 2-methyl-6-(phenylethynyl)-pyridine (MPEP) is a negative allosteric modulator, inhibiting the hydrolysis of phosphoinositides (Gasparini et al., 1999). MPEP can act as an inverse agonist of the mGlu5 receptor, inhibiting its constitutive activity (Gasparini et al., 1999; Pagano et al., 2000). In the same way, another potent selective mGlu5 receptor inhibitor is 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine (MTEP), which shows higher affinity for the receptor and higher solubility in water compared with MPEP (Cosford et al., 2003). Recent studies report that 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine (CTEP), a negative allosteric modulator with inverse agonist activity, is orally bioavailable and has in vivo affinity properties that are 30- to 100-fold higher than MPEP (Lindemann et al., 2011). Examples of mGlu5 receptor synthetic ligands are presented in Table 1.
Examples of mGlu5 receptor synthetic ligands
Endocannabinoids and the Cannabinoid Type 1 Receptor
The search for the mechanism of action of the main psychoactive component of the herb Cannabis sativa (hemp), Δ9-tetrahydrocannabinol, and its synthetic cannabinoid derivatives led to the discovery of the endogenous cannabinoid system (Mechoulam and Gaoni, 1967; Paton, 1975; Bisogno et al., 2005; Wang and Ueda, 2009). The endocannabinoid system comprises at least two endogenous ligands (endocannabinoids), the biochemical machinery for endocannabinoid synthesis and degradation, and the cannabinoid receptors, which include the cannabinoid type 1 (CB1) receptor, the cannabinoid type 2 receptor, and possibly others (Mechoulam et al., 1995; Piomelli, 2003; Di Marzo et al., 2004; Di Marzo, 2008). CB1, a Gi/o-coupled receptor, is abundantly expressed in the mammalian CNS and is mainly located at the presynaptic sites of neurons of the basal ganglia, cerebellum, hippocampus, certain regions of the cerebral cortex, ventral striatum, amygdala, periaqueductal gray (PAG) matter, and hypothalamic nuclei (Herkenham et al., 1990; Matsuda et al., 1990).
The major endogenous ligands of the cannabinoid receptors are arachidonoyl ethanolamide (AEA; also termed anandamide) and 2-arachidonoyl glycerol (2-AG) (Devane et al., 1992; Mechoulam et al., 1995). Besides AEA and 2-AG, other ligands have been proposed (Bisogno et al., 2000; Hanus et al., 2001; Huang et al., 2002; Porter et al., 2002). Unlike other mediators, AEA and 2-AG are not stored in the CNS, but they act as retrograde messengers that are synthesized and released in an activity-dependent manner, after changes in cellular homeostasis (Wilson and Nicoll, 2001; Marsicano et al., 2003; Di Marzo and Matias, 2005). Increased Ca2+ levels can augment endocannabinoid synthesis, in part because their biosynthetic enzymes are sensitive to Ca2+ concentration levels (Di Marzo, 2009).
Endocannabinoids are generally inactivated by mechanisms involving carrier-mediated transport into the cell and intracellular hydrolysis (Piomelli, 2003). Because AEA and 2-AG are lipophilic molecules, they can diffuse through the lipid membranes of both neurons and glia using carrier-mediated transport by facilitated diffusion (Beltramo et al., 1997; Hillard et al., 1997; Piomelli, 2003). AEA is metabolized into arachidonate and ethanolamine by the enzyme fatty acid amide hydrolase (FAAH) and 2-AG in arachidonate and glycerol via monoacylglycerol lipase (MAGL) (Piomelli, 2003; Di Marzo and Petrosino, 2007). Evidence also suggests that FAAH can catalyze 2-AG hydrolysis (Di Marzo and Deutsch, 1998; Bisogno et al., 2005). Since FAAH is present at cell bodies and dendrites of principal neurons and MAGL is located at axon terminals, it has been proposed that AEA and 2-AG degradation occurs at the post- and presynaptic neuron, respectively (Tsou et al., 1998; Dinh et al., 2002; Egertová et al., 2003).
CB1 Receptor: Pharmacology and Cell Signaling Pathways
CB1 receptor activation influences multiple effector systems; because of its wide distribution in the brain, the CB1 receptor is believed to be responsible for mediating the majority of the cannabinoids’ effects in the CNS (Cabral and Marciano-Cabral, 2005; Di Marzo, 2009). Evidence obtained by multiple groups supports the hypothesis that the CB1 receptor plays a key role in various cellular events (Ferreira-Vieira et al., 2014; Gomez et al., 2015; Zhang et al., 2015). Moreover, it has been reported that the CB1 receptor is involved in cognitive and affective processes, as well as in the control of motor activity, stress, addiction, and neuroprotection (Nagayama et al., 1999; Broadbent et al., 2004; Finn, 2010; Naydenov et al., 2014; McReynolds et al., 2016). Given its presynaptic location, it has been proposed that the CB1receptor can regulate neurotransmitter release and thus modulate synaptic plasticity by facilitating LTD (Wilson and Nicoll, 2001; Gerdeman et al., 2002; Marsicano et al., 2002; Robbe et al., 2002). The effect of CB1 receptor activation on synaptic plasticity can occur through the inhibition of presynaptic voltage-gated Ca2+channels, including N- and P/Q-type channels, or by the activation of A-type and inwardly rectifying K+ channels (Felder et al., 1993; Twitchell et al., 1997; Howlett et al., 2002; Wilson and Nicoll, 2002; Piomelli, 2003).
CB1 receptor stimulation activates multiple signal transduction pathways through the Gi/o family of G proteins (Fig. 2). Usually, CB1 receptor activation modulates both adenylate cyclase and mitogen-activated protein kinase (MAPK) activity. The classic effect of a CB1 receptor agonist is the inhibition of adenylate cyclase activity and reduction of cAMP production, which has been demonstrated in several conditions (Felder et al., 1993; Vogel et al., 1993; Childers et al., 1994). CB1 receptor agonists induce MAPK phosphorylation and MAPK activation, which suggests that the CB1 receptor can modulate gene transcription (Bouaboula et al., 1995; Patel et al., 1998). Furthermore, MAPK regulates key cellular processes (e.g., cellular proliferation and survival) that may be mediated by the CB1 receptor (Aguado et al., 2006, 2007). Other effects attributed to the CB1 receptor appear to involve activation of the PI3K/AKT pathway (Molina-Holgado et al., 2002; Ozaita et al., 2007). Activation of the anandamide/CB1/PI3K pathway was recently reported to protect rat brains from cocaine-induced neurotoxicity (Vilela et al., 2015).
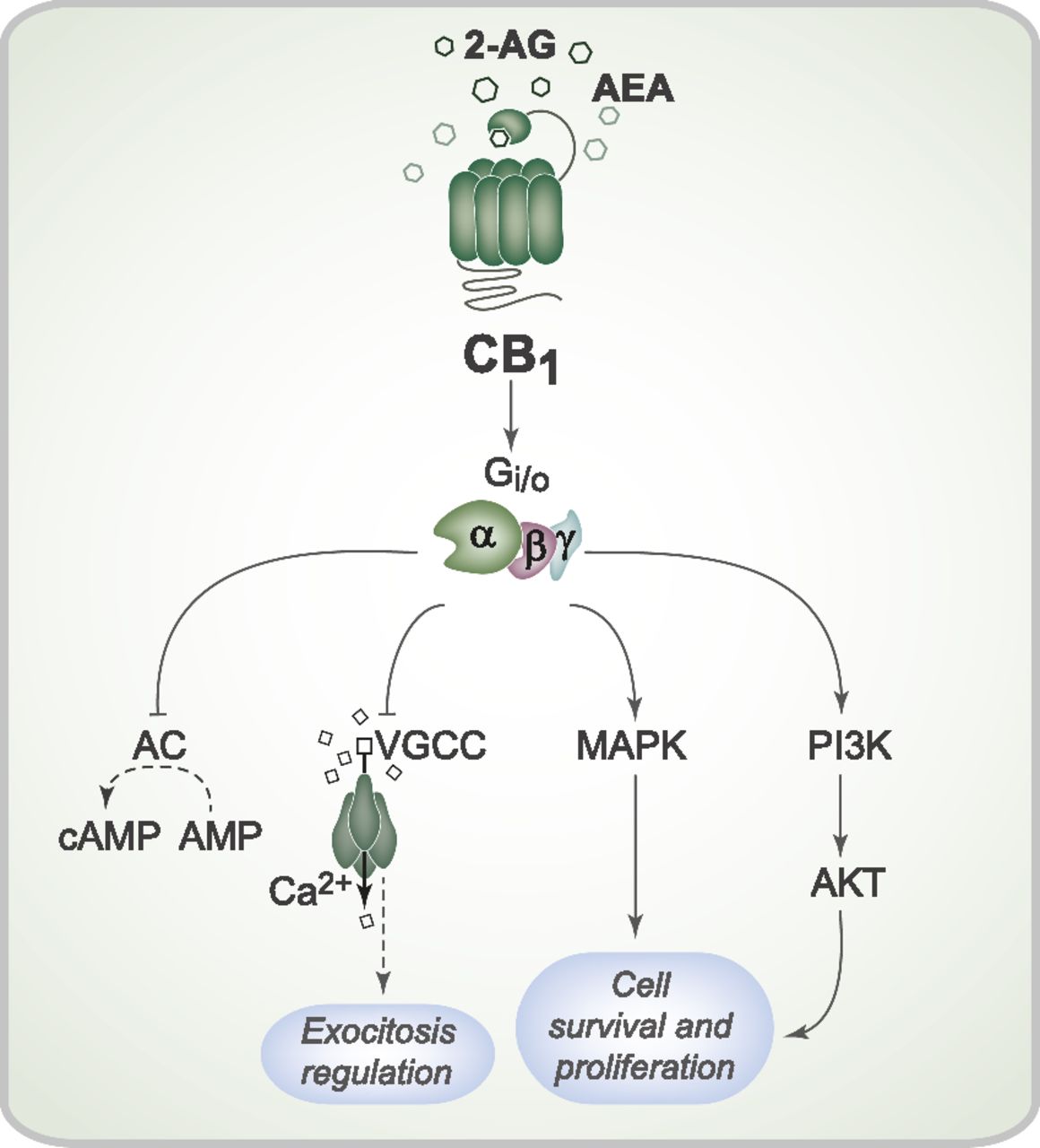
Cell signaling pathways triggered by CB1 receptor activation. Activation of the CB1 receptor by the endocannabinoids 2-AG and AEA can initiate a series of intracellular events mediated by the Gi/o protein. CB1 receptor activation can inhibit adenylate cyclase activity and therefore modulate cAMP production. CB1 receptor activation also promotes closure of the voltage-gated Ca2+ channel, which can reduce Ca2+ entry into the presynaptic neurons and interfere with synaptic vesicle exocytosis and neurotransmission. Furthermore, CB1 receptor stimulation can induce both MAPK and AKT phosphorylation and activation, which regulate proliferation and cell survival. AC, adenylate cyclase; VGCC, voltage-gated Ca2+ channel.
Some evidence indicates that pharmacological manipulation of the cannabinoid system can alleviate symptoms of several conditions, including addiction, seizures, neurodegenerative disease, and nociception (Adamczyk et al., 2009; Almeida-Santos et al., 2013; Pietropaolo et al., 2015; Vilela et al., 2015; Wolkers et al., 2015). For example, treatment with a CB1 receptor agonist, WIN 55,212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-4-morpholinylmethyl)pyrrolo[1,2,3,-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate], prevented the appearance of motor deficits, decreased the number of striatal huntingtin inclusions, and rescued striatal medium-sized spiny neurons loss in a Huntington disease mouse model (Pietropaolo et al., 2015). Positive results have also been found in Alzheimer and Parkinson disease mouse models (Price et al., 2009; Aso et al., 2012; Cao et al., 2014). Other synthetic compounds, termed indirect agonists, are of therapeutic value for their ability to potentiate the action of endogenous ligands in a controlled manner by blocking metabolism or uptake of endocannabinoids. For instance, we demonstrated that URB597 (cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester), an FAAH inhibitor, reduces cocaine-induced seizures and epileptiform electroencephalographic activity in rats (Vilela et al., 2015). Similarly, JZL-184 (4-nitrophenyl-4-[bis(1,3-benzodioxol-5-yl)(hydroxy)methyl]piperidine-1-carboxylate), an MAGL inhibitor, increases the levels of 2-AG, which reduces and delays the development of generalized seizures in a kindling model of temporal lobe epilepsy (von Rüden et al., 2015). Other effects, such as an increase in brain-derived neurotrophic factor levels, cellular proliferation, and neurogenesis, have also been reported with the use of cannabinoid receptors as indirect agonists (Ferreira-Vieira et al., 2014; Gomez et al., 2015; Zhang et al., 2015). See Table 2 for more examples of synthetic ligands of the endocannabinoid system.
Examples of synthetic ligands of the endocannabinoid system
The Functional Interaction between mGlu5 and CB1 Receptors
mGlu5 and CB1 receptors exhibit a close relationship, because one receptor can regulate the activation of the other (Fig. 3). CB1 receptor activation leads to decreased Ca2+ channel activity, which limits glutamate release and suggests that the CB1 receptor might regulate the amount of glutamate available to activate mGlu5 receptors (Shen et al., 1996; Wilson and Nicoll, 2001; Marsicano et al., 2003). On the other hand, mGlu receptor activation can facilitate endocannabinoid synthesis. For example, mGlu 1 receptor stimulation in cerebellar Purkinje cells can enhance endocannabinoid synthesis (Maejima et al., 2001). Moreover, stimulation of group I mGlu receptors, especially the mGlu5 receptor, in hippocampal CA1 cells also leads to increased cannabinoid production (Varma et al., 2001; Ohno-Shosaku et al., 2002). A rise in postsynaptic Ca2+ due to neuronal stimulation increases cannabinoid synthesis, which can be prevented when calcium chelators such as EGTA or 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid are present postsynaptically (Llano et al., 1991; Pitler and Alger, 1994). However, mGlu receptor activation by DHPG can augment the depolarization-induced release of endocannabinoids without increasing intracellular Ca2+levels (Ohno-Shosaku et al., 2002). Thus, it is possible that the mGlu5receptor might rely on other mechanisms to increase endocannabinoid synthesis. More recently, it has been demonstrated that the mGlu5 receptor is part of the 2-AG signalosome (Fig. 3), which was first identified at excitatory synapses of the ventral striatum and prefrontal cortex and was later found in other regions of the mammalian CNS (Katona et al., 2006; Mátyás et al., 2008; Nyilas et al., 2009; Jung et al., 2012). This supramolecular complex contains Homer proteins, which bind to the mGlu5 receptor and to the two key enzymes responsible for 2-AG synthesis, PLCβ and diacylglycerol lipase (DGL)-α (Katona et al., 2006; Jung et al., 2007, 2012). mGlu5 receptor stimulation leads to PLCβ activation and formation of DAG, which can be rapidly converted into 2-AG because of the close proximity of the enzyme DGLα. Because the 2-AG signalosome is restricted to excitatory synapses, this increase in 2-AG synthesis mainly underlies a suppression of presynaptic glutamate release that can be important for preventing excitotoxicity but can also decrease excitability (Lafourcade et al., 2007; Huang et al., 2008; Katona and Freund, 2008).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Functional interaction between mGlu5 and CB1 receptors: the signalosome complex. At glutamatergic synapses, mGlu5 receptor intracellular domains interact with Homer protein, which is a scaffold protein that is important for assembling the complex known as the signalosome. This signalosome complex, which is located at the perisynapse, brings the mGlu5 receptor into close proximity to the two key enzymes responsible for 2-AG synthesis, PLCβ and DGLα, because Homer proteins bind to the mGlu5 receptor, PLCβ and DGLα. Thus, mGlu5 receptor stimulation leads to the activation of PLCβ and formation of IP3 and DAG, which is metabolized to 2-AG by DGLα. 2-AG moves across the synaptic cleft and activates the CB1 receptor, located at the presynaptic terminal. Activation of CB1 inhibits the voltage-gated calcium channel, reducing the influx of Ca2+ and, consequently, reducing glutamatergic vesicle exocytosis. The result of mGlu5 and CB1 receptor signaling crosstalk is the regulation of postsynaptic neurons excitability. Activated elements are represented by continuous arrows, inhibition is represented by blocked arrows and reduced activity of the biologic process is represented by the dashed arrow. The protein complex depicted in the dashed area, named the signalosome complex, is located at the plasma membrane of the perisynaptic region. Glu, glutamate; iGluR, ionotropic glutamate receptor; VGCC, voltage-gated calcium channel; vGLUT, vesicular glutamate transporter 1.
CB1 and mGlu5 Receptors Mediate LTD
LTD is a type of synaptic plasticity that likely contributes to learning and memory and can be triggered by activation of NMDARs and mGlu receptors (Dudek and Bear, 1992; Fitzjohn et al., 1999; Kemp and Manahan-Vaughan, 2004). LTD can be induced by DHPG and it has been shown that the mGlu5 receptor is the key mGlu receptor to trigger LTD (Liu et al., 1993; Palmer et al., 1997; Huber et al., 2001). Notably, mGlu5 receptor stimulation can trigger LTD in both excitatory and inhibitory synapses by mobilizing endocannabinoids. It has been shown that repetitive activation of presynaptic glutamatergic fibers can induce mGlu-dependent LTD of hippocampal inhibitory synapses in the CA1 area via retrograde cannabinoid signaling (Chevaleyre and Castillo, 2003). In addition, mGlu5-dependent LTD via mobilization of endocannabinoids, especially 2-AG, also takes place at excitatory synapses in the visual and prefrontal cortex (Lafourcade et al., 2007; Huang et al., 2008) as well as in the Schaffer collateral pathway (Izumi and Zorumski, 2012). The mGlu5 receptor can also trigger LTD in a mechanism that is dependent on protein synthesis and might also involve the CB1 receptor (Weiler and Greenough, 1993; Huber et al., 2002). This type of mGlu5/CB1-induced long-term potentiation is further discussed in the section below on the role of CB1/mGlu5 in fragile X syndrome (FXS). The cooperative actions triggered by mGlu5 and CB1 receptors to induce LTD indicate that these two receptors might be important in various physiologic processes, including memory and addiction. Indeed, CB1 and mGlu5receptors appear to be involved in cocaine addiction, because a single exposure of mice to cocaine increases expression of Homer, reduces mGlu5receptor plasma membrane expression, and abolishes retrograde LTD mediated by endocannabinoids (Fourgeaud et al., 2004).
The Role of CB1 and mGlu5 Receptors in FXS
FXS, the most common cause of inherited intellectual disability, is caused by the silencing of the fragile X mental retardation 1 (FMR1) gene with consequent expression arrest of the fragile X mental retardation protein (FMRP), an RNA-binding protein important for translation regulation (Verkerk et al., 1991; Darnell et al., 2011). It has been demonstrated that the mGlu5receptor plays an important role in FXS (Bear et al., 2004; Michalon et al., 2012). Activation of group I mGlu receptors leads to protein synthesis–dependent establishment of LTD, which is enhanced in the absence of FMRP expression (Weiler and Greenough, 1993; Huber et al., 2002). Consistent with these studies, coupling between mGlu receptor activation and endocannabinoid mobilization is enhanced at CA1 GABAergic synapses of FMR1 knockout mice, leading to augmented endocannabinoid-mediated LTD and increased neuronal excitability (Zhang and Alger, 2010). Similarly, in the FMR1 knockout mice dorsal striatum, mGlu5-mediated endocannabinoid activity at GABAergic synapses is increased (Maccarrone et al., 2010).
At excitatory synapses of the frontal cortex and ventral striatum, FMRP deletion promotes disorganization of the 2-AG signalosome, marked deficits in mGlu5-mediated 2-AG release, and diminished endocannabinoid-dependent, protein synthesis–independent LTD (Jung et al., 2012). Homer binding to DGLα is altered at excitatory synapses of FMR1 knockout mice, which leads to misplacement of DGLα in the cell and disruption of the 2-AG signalosome, although no alterations in DGLα localization have been observed at inhibitory synapses (Jung et al., 2012). Thus, LTD can be differentially regulated by FMRP, depending on whether it takes place at excitatory or inhibitory synapses. Supporting this hypothesis, when Homer interactions were disrupted by a peptide that binds to long Homers, hippocampal slices from both wild-type and FMR1 knockout mice became more excitable due to enhanced mGlu5-dependent endocannabinoid mobilization at GABAergic synapses, although endocannabinoid mobilization by mGlu5 receptors was decreased at excitatory synapses (Tang and Alger, 2015). Interestingly, antagonism of CB1 receptor activity prevented the development of hyperexcitability in both FMR1 knockout and membrane-permeable Homer CT peptide models (Busquets-Garcia et al., 2013; Tang and Alger, 2015). However, pharmacological enhancement of 2-AG levels by JZL184 normalized endocannabinoid-mediated LTD and corrected key behavioral abnormalities in FMR1 knockout mice (Jung et al., 2012). Thus, it is currently unclear whether CB1 receptor blockage or activation could be beneficial in FXS.
mGlu5 and CB1 Receptors Cooperatively Modulate Nociception
The analgesic effect of cannabinoids in the CNS is mainly mediated by CB1receptor expression in neural substrates involved in nociceptive neurotransmission, including the spinal dorsal horn, PAG, dorsal raphe nuclei, and thalamic ventroposterolateral nucleus (Martin et al., 1995). Group I mGlu receptors can modulate nociceptive responses in the spinal cord, ventrobasal thalamus, dorsal raphe, amygdala, and PAG (Young et al., 1997; Hama, 2003; Palazzos et al., 2006). Importantly, it has been shown that mGlu5 and CB1 receptors exhibit a functional interaction to modulate nociception in some of these brain substrates.
The midbrain PAG is the most extensively studied supraspinal area involved in the modulation of nociception. Interestingly, it has been shown that the mGlu5 receptor is able to exert a tonic modulation of nociception promoted by microinjections of WIN 55,212-2 in the PAG, which increases nociceptive response latency in the plantar test (Palazzo et al., 2001). Moreover, under neuropathic pain conditions, PAG cannabinoid-induced antinociception requires downstream activation of the mGlu5 receptor (de Novellis et al., 2005; Palazzo et al., 2012). The mechanism underling mGlu5/CB1cooperation at the PAG to promote analgesia has been identified, because a previous study demonstrated that mGlu5 receptor stimulation in PAG slices leads to 2-AG mobilization and retrograde inhibition of GABAergic transmission, which produces analgesia (Drew et al., 2008). Corroborating these data, microinjection of DHPG into the dorsolateral PAG triggered the release of 2-AG and enhanced stress-dependent antinociception (Gregg et al., 2012). This effect was reversed by pharmacologically blocking DAGLα or reducing expression through small interfering RNA silencing (Gregg et al., 2012). Moreover, immunohistochemical analysis demonstrated that, at the PAG, the mGlu5 receptor colocalizes with DAGLα at the postsynaptic terminals in a location juxtaposed to the presynaptic localization of CB1receptors, which suggests that the postsynaptic mGlu5-DAGLα cascade can trigger retrograde 2-AG signaling in the PAG to promote analgesia (Gregg et al., 2012). Since the mGlu5 receptor is mostly present perisynaptically, spillover glutamate is the main endogenous mechanism to activate this receptor at the PAG region (Nusser et al., 1994; Drew et al., 2008). For example, neuronal inhibition of glutamate transporters in PAG slices enhanced mGlu5-mediated depression of GABAergic synaptic transmission through a CB1-dependent mechanism (Drew et al., 2008). In addition, capsaicin and anandamide can activate transient receptor potential vanilloid receptor 1 and induce substantial glutamate release in the ventrolateral PAG to stimulate mGlu5-dependent 2-AG mobilization and analgesia (Maione et al., 2006; Starowicz et al., 2007; Kawahara et al., 2011; Liao et al., 2011). Moreover, several analgesic neuropeptides can trigger 2-AG mobilization via mGlu5 receptor stimulation by exciting glutamatergic cell bodies. It has been shown that substance P, neurotensin, and cholecystokinin can stimulate neurokinin 1, neurotensin 1/2, and cholecystokinin 1 receptors, respectively, on glutamatergic somas. This triggers glutamate release and leads to activation of perisynaptic mGlu5 receptors with consequent PAG disinhibition through 2-AG mobilization (Drew et al., 2009; Mitchell et al., 2009, 2011). Future studies will be important to determine whether this PAG disinhibition mechanism contributes to the antinociceptive effects of intra-PAG injection of substance P, neurotensin, and cholecystokinin.
As in the case of PAG, a functional interaction between mGlu5 and CB1receptors to modulate nociception takes place at the spinal cord and amygdala. It has been shown that the antihyperalgesic effect of WIN55,212-2 in rats submitted to unilateral loose ligation of a sciatic nerve is mediated through an interaction between CB1 and mGlu5 receptors in the spinal cord dorsal horn (Hama and Urban, 2004). Further studies demonstrated that DGLα colocalizes with the mGlu5 receptor at the postsynaptic sites of nociceptive spinal synapses and also that the CB1 receptor is present at the presynaptic excitatory axon terminals (Nyilas et al., 2009). Moreover, intrathecal activation of mGlu5 receptor at the lumbar level evoked endocannabinoid-mediated, stress-induced analgesia through 2-AG mobilization, suggesting a key role for 2-AG–mediated retrograde suppression of nociceptive transmission at the spinal level (Nyilas et al., 2009). mGlu5 and CB1 receptors also work cooperatively to promote analgesia in an arthritis pain model through a mechanism that involves CB1-mediated increase in mGlu5 receptor function in the medial prefrontal cortex with consequent inhibition of abnormally enhanced amygdala output in pain (Ji and Neugebauer, 2014). In addition, fear-conditioning analgesia is facilitated by CB1 receptor activation in the basolateral amygdala via a mechanism that involves the modulation of mGlu5 and GABA receptor signaling (Rea et al., 2013). Thus, mGlu5 and CB1 receptors work cooperatively in various brain substrates to modulate nociception.
Conclusions
The crosstalk between mGlu5 and CB1 receptors is important for regulation of several physiologic events, including LTD and neurotransmitter release. Furthermore, the mGlu5/CB1 signalosome complex appears to play a major role in some conditions, such as nociception and FXS, which indicates that these G protein–coupled receptors are potential therapeutic targets. After the discovery of the functional interaction between mGlu5 and CB1 receptors, several studies have emerged aiming to elucidate the mechanisms involved in this process. Although it is clear that the formation of the signalosome containing mGlu5-Homer-PLCβ-DGLα is a key factor contributing to mGlu5-dependent mobilization of 2-AG and to the regulation of neurotransmission, more studies are needed to clarify the mechanisms and signaling pathways involved in this interaction.
Acknowledgments
The authors thank Roenick P. Olmo and Alvaro G. Ferreira for insightful comments that significantly contributed to figure design.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Olmo, Ferreira-Vieira, Ribeiro.
Footnotes
- Received March 21, 2016.
- Accepted June 16, 2016.
-
I.G.O. and T.H.F.-V. contributed equally to this work.
-
This research was supported by the Fundação de Amparo a Pesquisa do Estado de Minas Gerais and the Conselho Nacional de Desenvolvimento Científico e Tecnológico.
Abbreviations
- AEA
- arachidonoyl ethanolamide or anandamide
- AKT
- protein kinase B
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CB1
- cannabinoid type 1
- CDPPB
- 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide
- CHPG
- (RS)-2-chloro-5-hydroxyphenylglycine
- CNS
- central nervous system
- CTEP
- 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine
- DAG
- diacylglycerol
- DGL
- diacylglycerol lipase
- DHPG
- 3,5-dihydroxyphenylglycine
- ERK1/2
- extracellular signal-regulated kinase
- FAAH
- fatty acid amide hydrolase
- FMRP
- fragile X mental retardation protein
- FXS
- fragile X syndrome
- IP3
- inositol 1,4,5-triphosphate
- IP3R
- inositol 1,4,5-triphosphate receptor
- JZL-184
- 4-nitrophenyl-4-[bis(1,3-benzodioxol-5-yl)(hydroxy)methyl]piperidine-1-carboxylate
- LTD
- long-term depression
- MAGL
- monoacylglycerol lipase
- MAPK
- mitogen-activated protein kinase
- mGlu
- metabotropic glutamate
- MPEP
- 2-methyl-6-(phenylethynyl)-pyridine
- MTEP
- 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine
- NMDAR
- N-methyl-d-aspartate receptor
- PAG
- periaqueductal gray
- PDK1
- pyruvate dehydrogenase kinase 1
- PI3K
- phosphoinositide 3-kinase
- PLC
- phospholipase C
- 2-AG
- 2-arachidonoyl glycerol
- URB597
- cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester
- WIN 55,212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-4-morpholinylmethyl)pyrrolo[1,2,3,-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
