
![]()
NEURAL REGENERATION RESEARCH
May 2015,Volume 10,Issue 5
May 2015,Volume 10,Issue 5
PERSPECTIVE
Humans have been using Cannabis and its extracts for a few thousand years as a medicinal and recreational drug. However, the chemical component in Cannabis sativa, Δ9-tetrahydrocannabinol (Δ9-THC), an exogenous cannabinoid, remained unknown until it was isolated and identified as the main psychoactive ingredient (Gaoni and Mechoulam, 1964). Discovery of the cannabinoid Δ9-THC was a milestone in the field of cannabinoids, since this led to the unveiling of a new biological regulation system in our body, i.e., the endocannabinoid (eCB) system. The composition of the eCB system has been depicted by researchers in the cannabinoid field over the past three decades (Figure 1), which consists of endogenous cannabinoids (eCBs), G protein coupled cannabinoid receptors (CB1R and CB2R), the enzymes synthesizing and metabolizing eCBs, and transporters or carriers for eCBs (Di Marzo et al., 2015; Xu and Chen, 2015).
CB1R is widely expressed in neurons and astrocytes in the brain, while CB2R is present primarily in the immune system and microglial cells, the resident immune cells of the brain. There are some reports regarding neuronal expression of CB2R in the brain, but the expression is not as abundant as that of CB1R (Kano et al., 2009). The distribution of CB1R and CB2R in the brain determines their functional roles in cellular and molecular events and behaviors. Although several endogenous cannabinoids have been identi- fied, N-arachidonoyl ethanolamide (AEA or anandamide) and 2-arachidonoylglycerol (2-AG) are the two most-studied eCBs (Kano et al., 2009; Di Marzo et al., 2015). AEA was the first identified eCB, which is synthesized from N-arachidonoyl phosphatidylethanolamine (NAPE) by NAPE-specific phospholipase D (NAPE-PLD) and metabolized by fatty acid amide hydrolase (FAAH) to arachidonic acid (AA), precursor of prostaglandins. 2-AG was the second identified eCB, which is produced largely from diacylglycerol (DAG) by diacylglycerol lipase α and β (DAGLα/β) and primari- ly hydrolyzed by monoacylglycerol lipase (MAGL) to AA. 2-AG is also degraded by the enzymes α, β hydrolase 6 and 12 (ABHD6/12) and oxidized by cyclooxygenase 2 (COX-2). At present, the identities of eCB transporters or carriers are still controversial. 2-AG is the most abundant eCB and a full agonist for CB1R and CB2R, while AEA is a partial agonist for CB1R and CB2R and an agonist for the vanilloid receptor or transient receptor potential cation channel subfamily V member 1 (TRPV1), which is important for sensation of temperature and pain.
The abundance of CB1R expressed in the brain is similar to that of glutamate receptors. Notably, CB1R expressed in neurons preferentially resides on the presynaptic ends both at inhibitory GABAergic and excitatory glutamatergic synapses. This suggests that the primary role of the eCB system in regulation of brain function is through mediating or modulating synaptic transmission and plasticity (Kano et al., 2009; Castillo et al., 2012). While AEA may mediate synaptic transmission and other functions via CB1R/2 and TRPV1, accumulated information shows that 2-AG is a principal eCB signaling molecule in regulation of synaptic efficacy by acting on presynaptically expressed CB1R. The presence of MAGL and DAGLα/β at pre- and post-synaptic sites and the alterations in synaptic transmission and plasticity produced by pharmacological or genetic inhibition of DAGLα/β or MAGL provide strong evidence that 2-AG is a retrograde sig- naling molecule synthesized postsynaptically and inactivated presynaptically (Kano et al., 2009; Castillo et al., 2012). The release of GABA or glutamate is inhibited at GABAergic or glutamatergic synapses as a result of presynaptic CB1R activa- tion by 2-AG signaling. This may underlie short- (depolariza- tion-induced suppression of inhibition, DSI; and depolariza- tion-induced suppression of excitation, DSE) and long-term synaptic plasticity (inhibitory long-term depression, iLTD, and excitatory LTD, eLTD), implying that eCB signaling plays an important role in preventing excess synaptic activities, thereby retaining integrity and efficacy of synapses. This is particularly important in neurological diseases. However, eCB signaling also contributes to potentiation of glutamatergic synaptic transmission through the suppression of GABAergic synaptic release (Castillo et al., 2012; Xu and Chen, 2015), suggesting that while eCB signaling preferentially mediates inhibitory effects on both GABAergic and glutamatergic syn- aptic transmissions, it is also important for facilitating glutamatergic synaptic transmission by concurrent depression of inhibitory synaptic activity (Xu and Chen, 2015).
Homeostatic regulation of brain functions by endocannabinoid signaling
Humans have been using Cannabis and its extracts for a few thousand years as a medicinal and recreational drug. However, the chemical component in Cannabis sativa, Δ9-tetrahydrocannabinol (Δ9-THC), an exogenous cannabinoid, remained unknown until it was isolated and identified as the main psychoactive ingredient (Gaoni and Mechoulam, 1964). Discovery of the cannabinoid Δ9-THC was a milestone in the field of cannabinoids, since this led to the unveiling of a new biological regulation system in our body, i.e., the endocannabinoid (eCB) system. The composition of the eCB system has been depicted by researchers in the cannabinoid field over the past three decades (Figure 1), which consists of endogenous cannabinoids (eCBs), G protein coupled cannabinoid receptors (CB1R and CB2R), the enzymes synthesizing and metabolizing eCBs, and transporters or carriers for eCBs (Di Marzo et al., 2015; Xu and Chen, 2015).CB1R is widely expressed in neurons and astrocytes in the brain, while CB2R is present primarily in the immune system and microglial cells, the resident immune cells of the brain. There are some reports regarding neuronal expression of CB2R in the brain, but the expression is not as abundant as that of CB1R (Kano et al., 2009). The distribution of CB1R and CB2R in the brain determines their functional roles in cellular and molecular events and behaviors. Although several endogenous cannabinoids have been identi- fied, N-arachidonoyl ethanolamide (AEA or anandamide) and 2-arachidonoylglycerol (2-AG) are the two most-studied eCBs (Kano et al., 2009; Di Marzo et al., 2015). AEA was the first identified eCB, which is synthesized from N-arachidonoyl phosphatidylethanolamine (NAPE) by NAPE-specific phospholipase D (NAPE-PLD) and metabolized by fatty acid amide hydrolase (FAAH) to arachidonic acid (AA), precursor of prostaglandins. 2-AG was the second identified eCB, which is produced largely from diacylglycerol (DAG) by diacylglycerol lipase α and β (DAGLα/β) and primari- ly hydrolyzed by monoacylglycerol lipase (MAGL) to AA. 2-AG is also degraded by the enzymes α, β hydrolase 6 and 12 (ABHD6/12) and oxidized by cyclooxygenase 2 (COX-2). At present, the identities of eCB transporters or carriers are still controversial. 2-AG is the most abundant eCB and a full agonist for CB1R and CB2R, while AEA is a partial agonist for CB1R and CB2R and an agonist for the vanilloid receptor or transient receptor potential cation channel subfamily V member 1 (TRPV1), which is important for sensation of temperature and pain.
The abundance of CB1R expressed in the brain is similar to that of glutamate receptors. Notably, CB1R expressed in neurons preferentially resides on the presynaptic ends both at inhibitory GABAergic and excitatory glutamatergic synapses. This suggests that the primary role of the eCB system in regulation of brain function is through mediating or modulating synaptic transmission and plasticity (Kano et al., 2009; Castillo et al., 2012). While AEA may mediate synaptic transmission and other functions via CB1R/2 and TRPV1, accumulated information shows that 2-AG is a principal eCB signaling molecule in regulation of synaptic efficacy by acting on presynaptically expressed CB1R. The presence of MAGL and DAGLα/β at pre- and post-synaptic sites and the alterations in synaptic transmission and plasticity produced by pharmacological or genetic inhibition of DAGLα/β or MAGL provide strong evidence that 2-AG is a retrograde sig- naling molecule synthesized postsynaptically and inactivated presynaptically (Kano et al., 2009; Castillo et al., 2012). The release of GABA or glutamate is inhibited at GABAergic or glutamatergic synapses as a result of presynaptic CB1R activa- tion by 2-AG signaling. This may underlie short- (depolariza- tion-induced suppression of inhibition, DSI; and depolariza- tion-induced suppression of excitation, DSE) and long-term synaptic plasticity (inhibitory long-term depression, iLTD, and excitatory LTD, eLTD), implying that eCB signaling plays an important role in preventing excess synaptic activities, thereby retaining integrity and efficacy of synapses. This is particularly important in neurological diseases. However, eCB signaling also contributes to potentiation of glutamatergic synaptic transmission through the suppression of GABAergic synaptic release (Castillo et al., 2012; Xu and Chen, 2015), suggesting that while eCB signaling preferentially mediates inhibitory effects on both GABAergic and glutamatergic syn- aptic transmissions, it is also important for facilitating glutamatergic synaptic transmission by concurrent depression of inhibitory synaptic activity (Xu and Chen, 2015).
As illustrated in Figure 1, the eCB system homeostatically regulates brain functions in health and disease. Several lines of evidence indicate that eCB signaling is required in a variety of physiological functions from appetite, pain, ageing, learning and memory to depression and anxiety (Kano et al., 2009; Mechoulam and Parker, 2013; Maccarrone et al.,
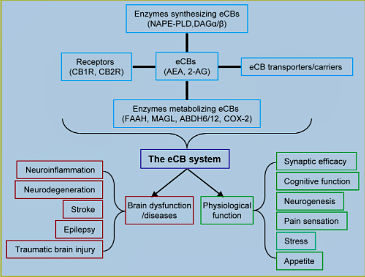
Figure 1 Homeostatic regulation of brain functions by the endocannabinoid (eCB) system in health and disease.
N-arachidonoyl ethanolamide (AEA) is synthesized by the enzyme N-acyl- phosphatidylethanolamine (NAPE)-specific phospholipase D (PLD) and inactivated by the enzyme fatty acid amide hydrolase (FAAH). 2-Arachidonoylglycerol (2-AG) is largely synthesized by the enzymes diacylglycerol lipase α (DAGLα) and DAGLβ and inactivated primarily by monoacyl- glycerol lipase (MAGL) and other enzymes, including α, β hydrolase 6 and 12 (ABHD6/12) and cyclooxygenase 2 (COX-2). Cannabinoid receptors (CB1R) is expressed in presynaptic terminals and astroglial cells. CB2R is primarily expressed in astroglial cells in the brain. The eCB system contrib- utes to normal physiological and neuropathological processes by homeostatic regulation of brain function.
2014; Di Marzo et al., 2015). For example, rimonabant (also known as SR141716), a CB1R receptor antagonist, was used for treatment of obesity by reducing appetite, although it was withdrawn from the market due to potentially undesirable psychological side effects. eCB signaling is also important for cognitive function. It has been demonstrated that working memory and extinction of aversive memory are impaired in animals administered with CB1R antagonists or agonists and in CB1R knockout mice (Mechoulam and Parker, 2013). Interestingly, elevation of endogenous 2-AG levels by MAGL inhibition enhances long-term potentiation (LTP) and promotes learning and memory (Chen et al., 2012; Xu and Chen, 2015). It has been reported that neurogenesis is reduced in mice deficient in DAGLα or β, and in CB1R or CB2 (Gao et al., 2010; Mechoulam and Parker, 2013). This suggests that neurogenes regulated by 2-AG signaling may contribute to synaptic plasticity and cognitive function (Xu and Chen, 2015). The involvement of 2-AG in synaptic plasticity is also supported from the study where high-frequency stimulation that induces LTP at hippocampal Schaffer col- lateral synapses elevates levels of 2-AG (Stella et al., 1997). In addition, a recent study provides evidence that eCB signaling may contribute to longevity in C. elegans (Folick et al., 2015). Therefore, the eCB system plays crucial roles in regulation of biological processes by maintaining homeostasis of brain functions. In particular, it responds differentially to patho- genic events or harmful assaults. For instance, the levels of 2-AG are significantly raised in the brain following traumat- ic brain injury (Panikashvili et al., 2001), while excitotoxic kainic acid exposure increases the levels of AEA, but not 2-AG (Marsicano et al., 2003). Additionally, the release of 2-AG, but not AEA, is elevated in response to brain β-amyloid (Aβ) infusion (van der Stelt et al., 2006). Perturbation of the eCB system may also lead to development of anxiety and depres- sion as well as other psychological disorders (Mechoulam and Parker 2013). Because of its widespread presence and functional roles in the brain, the eCB system displays profound anti-neuroinflammatory and neuroprotective properties. Neuropathological changes are alleviated by strengthening eCB signaling in several models of neurological diseases, including Alzheimer’s disease, traumatic brain injury, brain ischemia, seizures, Parkinson’s disease, and multiple sclerosis, which are closely associated with inflammation (Nomura et al., 2011; Chen et al., 2012; Mechoulam and Parker 2013; Di Marzo et al., 2015; Xu and Chen, 2015). Thus, manipulations of eCB signaling may provide novel pharmacotherapies for these intractable brain diseases.
Although the research in the past three decades has significantly advanced our understanding of the roles of the eCB system in physiological, pharmacological, and pathological processes, there are still a lot of unknowns about how these processes are regulated by eCB signaling at cellular, molecular, and genetic or epigenetic levels. It has been described that the formation and release of eCBs are ‘on demand’ in response to various events or stimuli. However, the precise mechanisms of how the release of eCBs occurs ‘on demand’ are still not clear. Previous studies show that the majority of physiological and pathological processes regulated by the eCB system are CB1R- or CB2R-dependent. Emerging evidence, however, suggests that many of these processes regulated by eCB signaling are through CB1R- or CB2R-in- dependent mechanisms, especially in counteracting of neu- ropathological events (Xu and Chen, 2015). The existence of a few orphan receptors such as G protein-coupled receptor 55 (GPR55) for eCBs indicates additional signaling pathways, including peroxisome proliferator-activated receptors (PPARs), in mediating biological functions of the eCB system. The complexity and diversity of the eCB system thus suggest that a broader spectrum of brain functions is ho- meostatically regulated by eCB signaling.
Although the research in the past three decades has significantly advanced our understanding of the roles of the eCB system in physiological, pharmacological, and pathological processes, there are still a lot of unknowns about how these processes are regulated by eCB signaling at cellular, molecular, and genetic or epigenetic levels. It has been described that the formation and release of eCBs are ‘on demand’ in response to various events or stimuli. However, the precise mechanisms of how the release of eCBs occurs ‘on demand’ are still not clear. Previous studies show that the majority of physiological and pathological processes regulated by the eCB system are CB1R- or CB2R-dependent. Emerging evidence, however, suggests that many of these processes regulated by eCB signaling are through CB1R- or CB2R-in- dependent mechanisms, especially in counteracting of neu- ropathological events (Xu and Chen, 2015). The existence of a few orphan receptors such as G protein-coupled receptor 55 (GPR55) for eCBs indicates additional signaling pathways, including peroxisome proliferator-activated receptors (PPARs), in mediating biological functions of the eCB system. The complexity and diversity of the eCB system thus suggest that a broader spectrum of brain functions is ho- meostatically regulated by eCB signaling.
This work was supported by National Institutes of Health grants NS076815.
*
Neuroscience Center of Excellence, School of Medicine, Louisiana State University Health Sciences Center, New Orleans, LA, USA
*
Neuroscience Center of Excellence, School of Medicine, Louisiana State University Health Sciences Center, New Orleans, LA, USA
Chu Chen
*Correspondence to: Chu Chen, Ph.D., cchen@lsuhsc.edu or chen502@gmail.com.
Accepted: 2015-04-03
doi:10.4103/1673-5374.156947
Accepted: 2015-04-03
doi:10.4103/1673-5374.156947
http://www.nrronline.org/
Chen C (2015) Homeostatic regulation of brain functions by endo-
Chen C (2015) Homeostatic regulation of brain functions by endo-
cannabinoid signaling. Neural Regen Res 10(5):691-692.
References
Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y (2012) Endocannabi- noid signaling and synaptic function. Neuron 76:70-81.
Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang YP, Teng Z, Chen C (2012) Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep 2:1329-1339.
Di Marzo V, Stella N, Zimmer A (2015) Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci 16:30-42.
Folick A, Oakley HD, Yu Y, Armstrong EH, Kumari M, Sanor L, Moore DD, Ortlund EA, Zechner R, Wang MC (2015) Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science 347:83-86.
Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, et al. (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogene- sis in diacylglycerol lipase knock-out mice. J Neurosci 30:2017-2024.
Gaoni Y, Mechoulam R (1964) Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc 86:1646-1647.
Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89:309-380.
Maccarrone M, Guzmán M, Mackie K, Doherty P, Harkany T (2014) Pro- gramming of neural cells by (endo)cannabinoids: from physiological rules to emerging therapies. Nat Rev Neurosci 15:786-801.
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W, Di Marzo V, Behl C, Lutz B (2003) CB1 cannabinoid receptors and on-demand defense against excito- toxicity. Science 302:84-88.
Mechoulam R, Parker LA (2013) The endocannabinoid system and the brain. Annu Rev Psychol 64:21-47.
Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF (2011) En- docannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 34:809-813.
Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E (2001) An endogenous cannabinoid (2-AG) is neuroprotec- tive after brain injury. Nature 413:527-531.
Stella N, Schweitzer P, Piomelli D (1997) A second endogenous cannabinoid that mediates long-term potentiation. Nature 388:773-778.
van der Stelt M, Mazzola C, Esposito G, Matias I, Petrosino S, De Filippis D, Mi- cale V, Steardo L, Drago F, Iuvone T, Di Marzo V (2006) Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci 63:1410-1424.
Xu JY, Chen C (2015) Endocannabinoids in synaptic plasticity and neu- roprotection. Neuroscientist 21:152-168.

References
Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y (2012) Endocannabi- noid signaling and synaptic function. Neuron 76:70-81.
Chen R, Zhang J, Wu Y, Wang D, Feng G, Tang YP, Teng Z, Chen C (2012) Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep 2:1329-1339.
Di Marzo V, Stella N, Zimmer A (2015) Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci 16:30-42.
Folick A, Oakley HD, Yu Y, Armstrong EH, Kumari M, Sanor L, Moore DD, Ortlund EA, Zechner R, Wang MC (2015) Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science 347:83-86.
Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, et al. (2010) Loss of retrograde endocannabinoid signaling and reduced adult neurogene- sis in diacylglycerol lipase knock-out mice. J Neurosci 30:2017-2024.
Gaoni Y, Mechoulam R (1964) Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc 86:1646-1647.
Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M (2009) Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89:309-380.
Maccarrone M, Guzmán M, Mackie K, Doherty P, Harkany T (2014) Pro- gramming of neural cells by (endo)cannabinoids: from physiological rules to emerging therapies. Nat Rev Neurosci 15:786-801.
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W, Di Marzo V, Behl C, Lutz B (2003) CB1 cannabinoid receptors and on-demand defense against excito- toxicity. Science 302:84-88.
Mechoulam R, Parker LA (2013) The endocannabinoid system and the brain. Annu Rev Psychol 64:21-47.
Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF (2011) En- docannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 34:809-813.
Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E (2001) An endogenous cannabinoid (2-AG) is neuroprotec- tive after brain injury. Nature 413:527-531.
Stella N, Schweitzer P, Piomelli D (1997) A second endogenous cannabinoid that mediates long-term potentiation. Nature 388:773-778.
van der Stelt M, Mazzola C, Esposito G, Matias I, Petrosino S, De Filippis D, Mi- cale V, Steardo L, Drago F, Iuvone T, Di Marzo V (2006) Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci 63:1410-1424.
Xu JY, Chen C (2015) Endocannabinoids in synaptic plasticity and neu- roprotection. Neuroscientist 21:152-168.