
 2020 Feb 14;10(1):2687. doi: 10.1038/s41598-020-59468-4.
2020 Feb 14;10(1):2687. doi: 10.1038/s41598-020-59468-4.Author information
Abstract
Radiotherapy combined with chemotherapy is the major treatment modality for human glioblastoma multiforme (GBM). GBMs eventually relapse after treatment and the average survival of GBM patients is less than two years. There is some evidence that cannabidiol(CBD) can induce cell death and increases the radiosensitivity of GBM by enhancing apoptosis. Beside initiation of death, CBD has been demonstrated as an inducer of autophagy. In the present study, we address the question whether CBD simultaneously induces a protective effect in GBM by upregulating autophagy. Addition of chloroquine that suppressed autophagic flux to 2D GBM cultures increased CBD-induced cell death, presenting proof for the protective autophagy. Blockage of autophagy upregulated radiation-induced cytotoxicity but only modestly affected the levels of cell death in CBD- or CBD/γ-irradiated 3D GBM cultures. Furthermore, CBD enhanced the pro-apoptotic activities of JNK1/2 and MAPK p38 signaling cascades while partially downregulated the pro-survival PI3K-AKT cascade, thereby changing a balance between cell death and survival. Suppression of JNK activation partially reduced CBD-induced cell death in 3D GBM cultures. In contrast, co-treatment of CBD-targeted cells with inhibitors of PI3K-AKT-NF-κB, IKK-NF-κB or JAK2-STAT3 pathways killed surviving GBM cells in both 2D and 3D cultures, potentially improving the therapeutic ratio of GBM.
Introduction
Human glioblastoma multiforme (GBM) is the most lethal primary brain cancer in adults. In the United States alone, approximately 12,000 patients are diagnosed with GBM each year1. The standard care for GBM includes surgical resection followed by concurrent external beam radiotherapy (with a total dose of 60 Gy) together with temozolomide (TMZ), as a supplemental DNA-methylating agent. It is followed by one year of adjuvant treatment with TMZ2. While external beam radiation therapy efficiently targets brain tumors, it could be accompanied by severe side effects, such as encephalopathy, strong cognitive and memory deficits3–8. Despite advances in GBM radiotherapy, outcomes remain poor with a median survival rate of 12–15 months after initial diagnosis9,10. In the past 10 years, survival in the treatment of this disease did not increase significantly. In spite of intense investigation on the gene drivers of GBM tumorigenesis, targeted therapies against these drivers have remained inefficient11.
Numerous studies in the recent years revealed pronounced cytotoxic effects of cannabinoids [cannabidiol (CBD) and Δ9-tetrahydrocannabinol (THC)] for GBM in cell culture and experimental animals12–16. Furthermore, preclinical studies demonstrated that treatment of GBM by combinations of cannabinoids and γ-irradiation or cannabinoids with TMZ was more effective against cancer cells than non-transformed cells in the brain17–19.
To further increase efficiency, specificity, and safety of the treatment, we address in the present study the question whether autophagy could play a protective role in the survival of CBD/γ-irradiation-treated GBM cells and, correspondingly, if upregulation of CBD-induced death can be enhanced by repression of the lysosomal/autophagic function in GBM cells. The key characteristic of macroautophagy (hereafter called autophagy) is the delivery of cytoplasmic cargo (including damaged organelles, protein aggregates, and even intracellular parasites) to the lysosomes for degradation to maintain nutrient homeostasis and organelle quality control20. Different forms of stress (including cell starvation, oxidative stress or, oppositely, a deficiency of oxygen and endoplasmic reticulum stress) could substantially upregulate the basal levels of autophagy providing cell survival functions21. A positive role of autophagy for cancer cell maintenance was observed at the beginning of stress conditions while a negative role was detected in situations when all life resources in the cells have already been used22,23. Consequently, many cancer cells, compared to normal cells, exhibit an increase in basal levels of autophagy that correlates with their higher survival during stress conditions. In contrast, senescent cells are characterized by the suppression of autophagy24,25. Interestingly, chemotherapy-induced senescent cells frequently restore phagocytosis and engulf both neighboring senescent or nonsenescent tumor cells at a remarkable frequency. Engulfed cells are processed through the lysosome and broken down, and cells that have engulfed others obtain a survival advantage, due to advanced nutrition conditions26.
Non-psychogenic CBD, a powerful inducer of intracellular oxidative stress15,27, was previously demonstrated as an initiator of both apoptotic and autophagic signaling pathways in several types of cancer cells including GBM12,28–30. A challenging problem is to further elucidate interference between apoptosis and autophagy during GBM cell death (especially, using of 3D GBM cultures) to find optimal and safe conditions for cancer treatment.
Results
CBD-mediated upregulation of autophagy in a 2D culture of U87MG human GBM cells
U87MG stock cultures from liquid nitrogen were continuously grown during several (8–10) passages in cell media supplemented with 10% FBS. Some experiments were performed with adherent U87MG cells grown as 2D monolayer cultures in cell media supplemented with 5% FBS that could promote autophagy. GBM cells were treated by CBD (5–20 μM) and γ-irradiation (5–10 Gy), as previously described28,31. A conventional surrogate biomarker of radiation-induced DNA damage, active Ataxia Telangiectasia Mutated (ATM) kinase (Ser1981-P), which played a crucial role as a sensor of double-strand brakes in genomic DNA and as the initiator of DNA repair after nuclear ionizing irradiation32, was detected by Western blotting 6 h after treatment. ATM activation was followed by phosphorylation/activation of p53, a master regulator of cell death and survival (Fig. 1a). On the other hand, CBD (20 μM) treatment induced oxidative stress15,27 that was accompanied by strong activation of Jun N-terminal Kinase 1/2 (JNK1/2) and followed by a notable increase in BECLIN-1 levels (an upstream protein of the autophagic cascade) in U87MG cells (Fig. 1a). CBD-induced upregulation of JNK1/2 activity is linked with i) transcriptional regulation of autophagic genes (including BECLIN1 with the corresponding increase of total protein BECLIN-1 levels)33 and ii) phosphorylation of BCL2 protein followed by releasing BECLIN-1 from a complex with BCL234. Furthermore, we observed a substantial increase in the levels of lipidated microtube-associated protein light chain 3-II (LC3-II) that reflected an increase in autophagosome maturation 6 h after CBD treatment (Fig. 1b). We additionally used ATM inhibitor (ATMi) KU6001935, alone or in combination with γ-irradiation and CBD, to investigate its downstream effects on autophagy. We previously confirmed specificity of ATM kinase inhibitor (ATMi) KU60019 (at concentration 1–2 μM) for suppression γ-irradiation-induced ATM activation in U87MG cells31. Interestingly, the presence of ATMi (1–2 μM) in non-irradiated or, especially, in γ-irradiated GBM cells was linked with upregulation of LC3-II levels 24–48 h after treatment (Fig. 1b), reflecting a role of ATM repression for increasing autophagic flux. Finally, confocal images demonstrated a substantial escalation of the cytoplasmic LC3 puncta formation after CBD (20 μM) treatment while γ-irradiation alone, besides modest effects on the cytoplasmic LC3 puncta formation, dramatically increased the nuclear LC3 levels (Fig. 1c,d). Cytoplasmic, perinuclear and nuclear localization of the LC3 puncta was previously observed in control and treated U87MG GBM cells36.

CBD induced autophagy in U87MG GBM 2D cell culture. (a) Western blot analysis of cell signaling proteins was performed 6 h after specified treatment of U87MG cells with CBD (20 μM) and γ-irradiation (10 Gy), alone or in combination. (b) Western blot analysis of LC3-II and LC3-I autophagy-related proteins was performed 6 h, 24 h and 48 h after treatments of GBM cells with CBD (20 μM), ATMi (2 μM) and γ-irradiation (10 Gy), alone or in combination. Original blots are shown in the “Supplementary information” section. After protein transfer, blot membranes were cut in two (or three) parts, which contained high molecular weight and low molecular weight proteins, respectively. The delineation of membranes was based on the well-known apparent molecular weight of investigated proteins. Cutting membranes were utilized for incubation with corresponding primary antibodies. The center lanes in LC3-I/II and β-ACTIN 24 h blots (which contain protein sample after an additional treatment non-used in this paper) were removed. (c) The LC3 puncta formation after indicated treatments of U87MG cells was detected using confocal microscopy with anti-LC3 Ab (green), anti-α-Tubulin Ab (red) and DAPI (blue). Images are shown with scale bar = 10 μm. (d) Relative levels of cytoplasmic green fluorescence (LC3) were determined using confocal images.
Effects of a triple combination of CBD, γ-irradiation, and chloroquine (CQ) on the viability of 2D U87MG GBM culture
CQ, a well-established clinical antimalarial drug that blocked the general lysosomal function and autophagosome fusion with lysosomes, was previously used for GBM treatment37,38. The presence of CQ also prevented LC3-II degradation by lysosomes in the cells and caused an additional increase in LC3-II/LC3-I ratio in the control and in CBD-treated cells (Fig. 2a). Eight hours after treatment, γ-irradiation alone or in combination with CBD, upregulated the autophagic flux in U87MG cells. At this time point, CBD also caused substantial phosphorylation/activation of both mitogen-activated protein kinase p38 (MAPK p38) and JNK that was correlated with increased levels of BECLIN-1 (Fig. 2a), while treatment with CQ (20 μM) alone upregulated only JNK activity and was not able to notably increase BECLIN-1 levels. CQ co-treatment resulted in blocking of autophagic flux for both treatment options: i) γ-irradiation alone or ii) γ-irradiation+CBD, which were characterized by the similar ratio of LC3-II/LC3-I (Fig. 2a). On the other hand, the presence of CBD was accompanied by a notable downregulation of AKT1-P levels in both non-irradiated and irradiated U87MG cells (Fig. 2a). Confocal microscopy demonstrated that CBD and CBD+CQ treatments resulted in an increase of the LC3 puncta formation in the cytoplasm and some upregulation of LC3 levels in the nuclei of U87MG cells, while γ-irradiation, besides an additional increase in cytoplasmic LC3, dramatically increased nuclear LC3 levels (Fig. 2b,c). A triple combination of CBD, γ-irradiation and CQ appears to induce redistribution of LC3 from the nuclei to the cytoplasm 8 h after treatment.

Effects of suppression of autophagic flux by chloroquine (CQ) on autophagy biomarkers and signaling proteins linked with autophagy regulation 8 h after treatment by CBD and γ-irradiation. (a) Western blot analysis of indicated signaling proteins from U87MG cells was performed 8 h after treatments with CQ (20 μM) in the presence of 0.1% DMSO (no CBD) or CBD (20 μM) diluted in 0.1% DMSO in non-irradiated and γ-irradiated (10 Gy) cells. Original blots are shown in the “Supplementary information” section. After protein transfer, blot membranes were cut in two (or three) parts, which contained high molecular weight and low molecular weight proteins, respectively, and utilized for incubation with corresponding primary antibodies. (b,c) The LC3 puncta formation after indicated treatments of U87MG cells was detected using confocal microscopy with anti-LC3 Ab, anti-α-Tubulin Ab, and DAPI. Scale bars = 10 μm. (d,e) Relative U87MG cell viability (based on the intracellular ATP level) was determined 24–48 h after treatments with CBD (10–20 μM) and CQ (10–20 μM) alone or in combination, without or with γ-irradiation (10 Gy). CBD (diluted in 0.1% DMSO), as well as control 0.1% DMSO were added 30 min before irradiation, CQ (10–20 μM) was added 15 min before irradiation. Pooled results of four independent experiments using U87MG cells 24–48 h after treatments are shown. Error bars represent means ± S.D. (p < 0.05, Student’s t-test). The stars and arrows indicate significant differences between the specified treatments of cancer cells.
The effects of CBD, CQ, and γ-irradiation (10 Gy), alone or in combination, on the viability of 2D U87MG cultures 24–48 h after treatment were examined (Fig. 2d,e). We observed slight-to-moderate effects of individual or combined treatments after 24 h and very strong negative effects on GBM cell viability 48 h after treatment with CBD (20 μM), CBD (20 μM) + 10 Gy and, especially, CBD (20 μM) + CQ (20 μM) + 10 Gy. The presence of CQ (20 μM), indeed, exhibited additional negative effects on cancer cell survival at time points, 24 h and 48 h (Fig. 2d,e), highlighting some protective effects of autophagy in this scenario.
Dose-dependent effects of CQ in combination with CBD on cell signaling pathways and cell death regulation in 2D GBM cell cultures
We further elucidated dose-dependent effects of CQ (10–40 μM) alone or in combination with CBD (20 μM), on the main cell signaling pathways that regulate cell survival and death 24–48 h after treatment in U87MG cells (Fig. 3a). CBD and CQ, alone or in combination, upregulated JNK-phosphorylation/activation, however, cJUN-P levels increased only in the presence of CBD. A critical feature of CBD (20 μM), a strong suppression of AKT-1 phosphorylation/activation, was undoubtedly revealed 24 h after treatment and was not additionally affected by CQ (10–40 μM) (Fig. 3a). It was accompanied by a significant downregulation in levels of the critical downstream targets of AKT-1, mammalian Target of Rapamycin (mTOR)-P(S2448) and β-Catenin-P(S552) (Fig. 3a). At this time point, CBD caused a moderate downregulation of Extracellular signal-Regulated Kinase 1/2 (ERK1/2) and Nuclear Factor kappa B (NF-κB) p65 phosphorylation that was also well-pronounced for a combination of CBD and CQ. In general, we observed a notable suppression of cell survival pathways [such as Phosphatidylinositol 3-kinase (PI3K)-AKT/NF-κB or PI3K-AKT-mTOR] and promotion of JNK-mediated pathways (that also regulate autophagic and apoptotic signaling) by CBD (20 μM) alone or in combination with CQ. The presence of CQ also caused a mild downregulation of basal levels of Hypoxia Induced Factor 1-alpha (HIF-1α) in U87MG cells (Fig. 3a). Furthermore, a combination of CBD and CQ demonstrated specific effects on modulation of autophagic flux, such as an increase in LC3-II/LC3-I ratio for CBD (20 μM) with an additional increase in the presence of CQ (20–40 μM).

Dose-dependent changes in the pattern of cell signaling proteins of 2D U87MG GBM cell cultures and cell viability 24–48 h after treatment with CQ alone or in combination with CBD. (a,b) Western blot analysis of indicated signaling proteins from U87MG cells was performed 24 h (panel a) and 48 h (panel b) after treatments with CQ (10–40 μM) in the presence of 0.1% DMSO (no CBD) or CBD (20 μM) diluted in 0.1% DMSO. Original blots are shown in the “Supplementary information” section. After protein transfer, blot membranes were cut in two (or three) parts, which contained high molecular weight and low molecular weight proteins, respectively, and utilized for incubation with corresponding primary antibodies. (c) Relative viability of U87MG and T98G GBM cells (based on intracellular ATP levels) 24–48 h after indicated treatment. Error bars represent means ± S.D. (p < 0.05, Student’s t-test). The black and white arrows, the black and white circles, the stars and double stars indicate a significant difference in control and treated cells. (d) U87MG cells were fixed/permeabilized in 70%-ethanol and cell DNA was stained by PI. Cell cycle-apoptosis analysis was performed using flow cytometry. The gates M1 and M2 showed % of pre-G1 (late apoptotic cells) and G1 cells, respectively. The gate M3 contained cells of S- and G2/M phases of the cell cycle. A ratio G2/G1 of cell subpopulations was also determined using the algorithm of the FCS Express program.
On the other hand, downregulation of CBD-induced active (cleaved) caspase-9 and caspase-3 levels was observed for the combination of CBD (20 μM) and CQ (20–40 μM), indicating a partial repression of CBD-induced apoptotic signaling in these conditions (Fig. 3b). CBD, CQ and their combination demonstrated pronounced cytotoxic effects on U87MG accompanied by cell death, quantitative assessment of which was performed using a cell viability assay (Fig. 3c). This assay revealed a strong inhibition of U87MG cell viability by CBD (20 μM) that was additionally decreased in the presence of increasing doses of CQ 24–48 h after treatment. CQ alone caused dose-dependent but only a partial decrease in viability of a 2D culture of U87MG cells (Fig. 3c). We additionally performed cell viability assay for equally treated T98G and U118MG human GBM lines and revealed a similar pattern of negative effects of the combination of CBD and CQ for the survival of U87MG, T98G and U118MG cell lines grown as 2D cultures (Fig. 3c and data not shown). Furthermore, cell cycle-apoptosis analysis demonstrated substantial changes in the U87MG cell cycle by increasing doses of CBD (0–20 μM) resulting in an increase of G2/G1 ratio, while a ratio of the pre-G1 cell subpopulation was relatively stable 24 h after treatment (Fig. 3d). The presence of CQ (20 μM) notably increased a pre-G1 ratio, correlating with induction of cell death, probably, via late apoptosis and necrosis. Finally, CBD at a 40 μM dose demonstrated dramatic level of cytotoxicity for cancer cells even 24 h after treatment; the presence of CQ in these conditions did not further affected cell death (Fig. 3d).
Effects of CBD, γ-irradiation, and CQ on survival/death of U87MG GBM cells, TIG3 normal human embryonic fibroblasts and immortalized human fetal astrocytes (IHFA)
Two human non-malignant cell lines, TIG3 and IHFA, were used to elucidate cytotoxic effects of CBD, CQ and γ-irradiation on normal cell growth in 2D culture (Fig. 4). Annexin-V-FITC and PI staining and the subsequent flow cytometry demonstrated minor levels of total cell death (most necrotic) that was induced by CBD and CQ in TIG3 cells. Irradiation (10 Gy) induced additional levels of cell death in TIG3 culture (Fig. 4a–c). Treatment by CBD alone (20 μM) caused only modest morphological changes in TIG3 cells (Fig. 4e). Furthermore, CBD and CQ treatment resulted in minor cytotoxicity for a 2D culture of IHFA 48 h after treatment that was additionally increased by γ-irradiation (Fig. 4d). Taken together, these data indicated lower sensitivity of normal non-malignant cells to combined treatment with of CBD, γ-irradiation, and CQ, compared to adherent U87MG cells: the viability 48 h after treatment with CBD (20 μM), CQ (20 μM) and 10 Gy was around 60% for TIG3 cells, 80% for IHFA and less than 5% for U87MG (Figs. 4c,e and and2e).2e). Previously, we demonstrated a strong CBD-mediated apoptosis in U87MG and other GBM lines28.

Levels of cell death induced by CBD and CQ in U87MG cells, TIG3 (normal human embryonic fibroblasts) and (IHFA) immortalized human fetal astrocytes. (a–d) Annexin-V-FITC and PI staining for determination of the early apoptotic (EA), late apoptotic (LA) and secondary-necrotic (SN) cells after indicated treatment was followed by the flow cytometry. Typical experiment for TIG3 cells (panel a) and pooled results of four independent experiments (panels b–d) using (early apoptotic + late apoptotic + secondary necrotic) 48 h after indicated treatments of U87MG, TIG3 and IHFA are shown. Error bars represent means ± S.D. (p < 0.05, Student’s t-test). (e) The images of control and CBD-treated TIG3 cells after immunostaining with α-Tubulin and DAPI are shown. Scale bar = 20 μm. (f,g) CBD-mediated control of STAT3 activation in U87MG cells. Western blot analysis of STAT3-P(Y705), STAT3-P(S727) and total STAT3 4 h after indicated treatments are shown. Original blots are shown in the “Supplementary information” section. Only cut blot membranes with transferred protein in the range of 60–130 kD were used for incubation with antibodies to total or phosphorylated STAT3. (h) Cell viability assay of U87MG cells 48 h after specified treatments including CBD (20 μM), AG490 (40 μM) and STAT3 inhibitor-6 (40 μM). Error bars represent means ± S.D. (p < 0.05, Student’s t-test). Stars and circles indicate a significant difference in cell viability after indicated treatments.
CBD-mediated activation of signal transducer and activator of transcription 3 (STAT3) in U87MG cells
Beside CBD-induced JNK-cJUN/AP1 activation and permanently active NF-κB p65-p50 in several GBM lines (including U87MG), we elucidated CBD-mediated activation of STAT3, a crucial transcription factor that together with AP1/cJun and NF-κB regulates transcription of numerous targeted genes, including cytokines, autophagic biomarkers, pro-inflammatory enzymes (such as COX2) and anti-apoptotic proteins (BCL2 and BCL-XL)39,40. We observed substantial levels of total STAT3 protein expression in U87MG (Fig. 4f,g). CBD treatment after 4 h substantially upregulated STAT3 Tyr705-phosphorylation and notably STAT3 Ser727-phosphorylation that was crucial for the nuclear translocation and transcriptional activation of STAT3. As expected, Tyr705-phosphorylation of STAT3 was partially suppressed by AG490, a molecule inhibitor of Janus Kinase 2 (JAK2) (Fig. 4f). Permanently active NF-κB p65 was not notably affected 4 h after this treatment (not shown). We additionally utilized STAT3 inhibitor-6 (which suppressed STAT3 dimerization and nuclear translocation) for treatment CBD-targeted GBM cells that also resulted in a partial inhibition of STAT3-phospho-Tyr705 levels (Fig. 4g). Combined treatment by CBD (20 μM) and AG490 (20 μM) or by CBD and STAT3 inhibitor-6 (40 μM) resulted in an additional downregulation of survival of U87MG cells (Fig. 4h) highlighting a role of the JAK2-STAT3-P pathway as a general pro-survival factor in GBM cells before and after CBD treatment.
Effects of CBD alone or in combination with γ-irradiation and CQ on 3D U87MG GBM cell cultures
A 3D cancer cell culture is a more adequate model for the presentation of tumor architecture and for investigation of cancer treatment options, compared to 2D culture41,42. U87MG cells could be relatively easily established as 3D spheroid culture using serum-free media, with the necessary growth factor supplements (see “Methods”). We used two different 3D models for this cell line: i) spheroids (neurospheres) were grown as suspension cultures, ii) microtumors were grown in Matrigel-collagen matrices (Fig. 5a,c).

Treatment of 3D U87MG cultures by CBD, CQ, and γ-irradiation, alone or in combination. (a,b) U87MG spheroids grown in serum-free media with growth factors (see “Methods” section) were treated, as indicated. For confocal imaging, cells were stained CellTracker (red) (a); cell viability assay was performed in non-stained cells using the luminescent detection of ATP levels (b). (c,d) 3D U87MG cells were grown as a microtumor in a Matrigel – collagen matrices for 3–6 days after indicated treatments: 0.1% DMSO as a vehicle, CBD (5–10 μM in DMSO) and CQ (40 μM), with or without γ-irradiation. Cells were incubated 30 min with CBD or CBD+CQ before γ-irradiation (at 10 Gy). After the subsequent incubation for 72 h or 144 h, cells were labeled with CellTracker (red) and analyzed using fluorescent microscopy. The relative number of surviving cells was detected.
We investigated the cell viability of 3D suspension cultures after treatment with CBD, CQ, and γ-irradiation, alone or in combination. These cultures that enriched by glioma-initiating cells are known as radioresistant43. In our experiments, individual spheroids from the U87MG control suspension cultures contain 20–50 cells. After 48 h of CBD (20 μM) treatment, spheroids with a diffusible morphology appeared indicative of dead and dying cells (Fig. 5a). Contrarily, CQ caused spheroid fusion and, finally, treatment with combination of CBD and CQ partially suppressed effects of CBD on spheroid morphology (Fig. 5a). However, spheroid morphology (as shown in Fig. 5a) was not enough to determine levels of cell death. Quantitative cell viability assay (Fig. 5b) demonstrated strong cytotoxic effects of CBD, alone or in combination with γ-irradiation, on suspension cultures. The presence of CQ, an inhibitor of autophagic flux, did not accelerate CBD-induced death of neurospheres (Fig. 5b), indicating modest (or neutral) effects of autophagy in this scenario. Furthermore, due to a strong killing efficiency of CBD (20 μM) for 3D spheroid culture, 2–3 days after treatment (Fig. 5a,b) additional usage of γ-irradiation also resulted in a relatively slight increase in cell death. However, a combination of γ-irradiation and CQ was more cytotoxic, than γ-irradiation alone (Fig. 5b).
On the other hand, in the case of microtumors grown in Matrigel-collagen matrices using the serum-free media, the process of cell death induced by CBD at lower concentration (5–10 μM) was relatively slow (up to 6 days), compared to suspension culture, but well-pronounced, using cells labeling by CellTracker (red) and fluorescent microscopy accompanied by cell counting (Fig. 5c). Radiation pre-treatment (10 Gy) caused pronounced acceleration of cell death in this 3D model (that was not enriched with radioresistant glioma-initiating cells), especially, in combination with CBD (10 μM) (see Fig. 5c,d). CQ co-treatment together with CBD and γ-irradiation of Matrigel-grown 3D U87MG culture produced only slight additional effects on cell survival (Fig. 5c,d).
Effects of combined treatment of CBD and CQ on 3D TS543 proneural GBM spheroids
Despite strong evidence connecting several receptor tyrosine kinases, including the platelet-derived growth factor receptor (PDGFR), in the pathogenesis of GBM, the clinical use of receptor tyrosine kinases inhibitors for treatment of different forms of GBM has been greatly compromised by the rapid emergence of therapeutic resistance44. To overcome the resistance of proneural TS543 GBM that was driven by a PDGFR-regulated pathway to targeted tyrosine kinase inhibitors, we used CBD treatment of TS543 3D culture. In our recent study, we demonstrated effective killing of proneural TS543 GBM cells grown as 3D spheroid culture by CBD31. Additional investigation of the main signaling pathways linked with the autophagy regulation in TS543 cells after relatively short (3 h) treatment by CBD (5–20 μM) revealed, as expected, a strong dose-dependent activation/phosphorylation of JNK1/2 and MAPK p38. Interestingly, ERK2, cJUN and Activating Transcription Factor 2 (ATF2) phosphorylation/activation were maximal after treatment with CBD (10 μM) but not CBD (20 μM), probably due to initiation of cell death by CBD (at 20 μM). A modest decrease in basal p65-P NF-κB and AKT1-P levels was also detected 3 h after treatment with CBD (20 μM) (Fig. 6a). A prolonged (24 h) treatment of TS543 spheroids by CBD (10 μM), alone or, especially, in combination with CQ (20 μM) (see Fig. 6b) strongly suppressed p65-P(S536) NF-κB and AKT1-P levels but still upregulated JNK1/2-P and p38-P activities, as well as their targets cJUN-P and ATF2-P. In contrast, ERK2-P activity was strongly repressed by CQ but was present at a moderate level after treatment with CBD+CQ. Furthermore, treatment of TS543 spheroids by CBD, CQ, and CBD+CQ demonstrated a substantial increase in LC3-II/LC3-I, revealing pronounced effects on modulation of the autophagic flux (Fig. 6b). CBD (10–20 μM) alone demonstrated dramatic negative effects on spheroid viability 48 h after treatment, which was additionally downregulated in the presence of CQ (5–40 μM) (Fig. 6d). In general, additional effects of CQ on CBD-induced cell death were significant only for low-to-average (5–10 μM) doses of CBD (Fig. 6d). Substantial changes in the morphology of TS543 spheroids showing their damage and cell death after indicated treatments were demonstrated by the fluorescent microscopy of TS543 cells labeled by CellTracker (green) (Fig. 6c). Results obtained indicate efficient killing of TS543 proneural spheroids by CBD treatment with additional modest effects of CQ and ionizing radiation.

The viability of TS543 human proneural GBM spheroid cultures following treatments with CBD and CQ, alone or in combination. (a,b) Western blot analysis was performed to elucidate the dose-dependent response of TS543 spheroids to CBD (5–20 μM) 3 h after treatment or to CBD (10 μM) and CQ (20 μM), alone or in combination 24 h after treatment. Original blots are shown in the “Supplementary information” section. After protein transfer, blot membranes were cut in two (or three) parts, which contained high molecular weight and low molecular weight proteins, respectively, and utilized for incubation with corresponding primary antibodies. (c) CBD-treated (24 h) and control (0.1% DMSO treated) TS543 spheroids were additionally incubated with CellTracker (green) for 30 min and analyzed using fluorescent microscopy. Scale bars = 200 μm. (d) Combined treatment of TS543 spheroids using CBD (5–20 μM) and CQ (10–40 μM). Cell viability assay was performed for spheroid cultures 24–48 h after indicated treatments. Pooled results of four independent experiments 24–48 h after treatments are shown. Error bars represent means ± S.D. (p < 0.05, Student’s t-test). Circles, stars, and dual stars indicate significant differences in cell viability after specified treatments.
Effects of the suppression of JNK and AKT activities on levels of CBD-induced cell death in TS543 spheroids
CBD was shown to induce JNK activation as well as to suppress AKT1 activity in TS543 spheroids (Fig. 6a,b). To determine if these effects modulate the viability of TS543 spheroids upon treatment with CBD, we utilized small molecule inhibitors of JNK activity (SP600125) and PI3K-AKT (LY294002) together with a relatively low dose of CBD (5 μM). CBD-induced functional activity of JNK (determined by levels of cJUN phosphorylation) was effectively suppressed by SP600125 while its secondary effect on AKT1 phosphorylation was modest. Total levels of cJUN in the presence of SP600125 were also significantly decreased (Fig. 7a), probably, as a result of cJUN-P-dependent transcription of the cJUN gene45 and upregulation of the proteasome-dependent degradation of cJUN protein in the presence of non-active JNK46. On the other hand, LY294002 downregulated basal AKT1-P levels and further decreased CBD-affected AKT1-P levels (Fig. 7a). Determination of the cell viability 48–72 h after treatment confirmed, as expected, a protective role of suppression of CBD-induced JNK activation by SP600125 (that notably reduced levels of cell death after treatment with low doses of CBD). In contrast, inhibition of AKT1 activity additionally accelerated CBD-induced cell death (Fig. 7b,c). CBD, CBD+SP600125, and CBD+LY294002 also demonstrated their effects on LC3-II/LC3-I ratio that characterized the intensity of the autophagic flux. Fluorescent microscopy of TS543 spheroids (labeled with CellTracker green) further revealed opposite effects of SP600125 and LY294002 on the native structure and survival of TS543 spheroids (Fig. 7d).

TS543 spheroid culture following treatment by CBD and small molecular inhibitors of JNK (SP600125) and PI3K-AKT (LY294002), alone or in combination. (a) Western blot analysis was performed to elucidate the dose-dependent response of indicated signaling proteins in TS543 spheroids to CBD (5 μM) alone or in combination with SP600125 (20 μM) or LY294002 (25 μM) 24 h after treatment. Original blots are shown in the “Supplementary information” section. After protein transfer, blot membranes were cut in two parts, which contained high molecular weight and low molecular weight proteins, respectively, and utilized for incubation with corresponding primary antibodies. (b,c) Combined treatment of TS543 spheroids using indicated combinations of CBD (5–20 μM), SP600125 (20 μM) and LY294002 (25 μM). Cell viability assay was performed for spheroid cultures 48–72 h after indicated treatments. The pooled results of four independent experiments are shown. Error bars represent means ± S.D. (p < 0.05, Student’s t-test). Circle, dual circles, triple circles, stars and dual stars indicate significant differences in cell viability after specified treatments. (d) TS543 spheroids were treated, as indicated for 72 h and then labeled by incubation with CellTracker (green) for 30 min and analyzed using fluorescent microscopy. Scale bars = 200 μm.
Combined inhibition of the PI3K-AKT and AKT/IKK-NF-κB signaling pathways exaggerates CBD-induced death of TS543 spheroids
Beside PI3K-AKT, the Inhibitor Nuclear Factor kappa B Kinase (IKK)-NF-κB signaling pathway is permanently active in many GBM, including TS543 spheroids, and could be partially (but cell-specifically) suppressed by CBD (10–20 μM) treatment. To further elucidate a role of individual signaling pathways, including the NF-κB-dependent pathway, that could accelerate effects of CBD, we used co-treatment of CBD-targeted TS543 spheroids with small molecule inhibitors of several signaling pathways: PI3K-AKT-NF-κB pathway (LY294002), IKKβ-NF-κB (BMS345541), IKKα-IKKβ–NF-κB (Bay11-7082), MAPK p38 and AKT1 activities (SB203580) and JAK2-STAT3 (AG490). LY294002 (at 40 μM) completely suppressed the basal and CBD-targeted AKT-phosphorylation (Fig. 8a), compared to its partial effects at a dose of 25 μM (see Fig. 7a). NF-κB p65 phosphorylation/activation was markedly reduced by a combination of small molecule inhibitors of IKKβ (BMS345541 at 10 μM) and PI3K-AKT (LY294002 at 40 μM), because both pathways were involved in IκBα phosphorylation-degradation in GBM cells28. Furthermore, Bay11-7082, a IKKα irreversible inhibitor, also demonstrated NF-κB suppression (Fig. 8a,b). SB203580 (20 μM), a specific inhibitor of MAPK p38 activity, is also known as an AKT1 inhibitor at increased concentrations. Consequently, a combination of CBD+SB203580 suppressed AKT1 activity (Fig. 8a). Furthermore, CBD, alone or in specified combinations, suppressed β-Catenin phosphorylation linked with many critical cell functions. These treatment modalities were also involved in autophagic signaling in spheroids, as shown by an increase of LC3-II/LC3-I ratio (Fig. 8a). Cell viability assay demonstrated dramatic effects of CBD, especially, in a combination with inhibitors of NF-κB and PI3K-AKT signaling pathways on GBM cell survival, while an additional effect of SB203580 (alone) or in combination with CBD for GBM survival was relatively modest (Fig. 8c). AG490 (JAK2-STAT3 inhibitor) demonstrated in combination with CBD additive effects for decreasing TS543 viability (Fig. 8d). On the other hand, Rapamycin (mTOR inhibitor, a classical activator of autophagy) demonstrated a protective effect against CBD-induced death only at low-to-average CBD dose (5–10 μM) (Fig. 8d). Analysis of fluorescent images of TS543 spheroids (Fig. 8e) further confirmed strong damaging effects of CBD in concert with NF-κB/AKT suppression, allowing achievement of a dramatic decrease in cell viability and, possibly, to substitute the necessity of CQ pretreatment.

TS543 spheroid culture after treatment by CBD and small molecular inhibitors of PI3K-AKT (LY294002), MAPK p38 (SB203580), IKKb (BMS344551), IKKa (Bay11-7082), JAK2 (AG490) and mTOR (Rapamycin) alone or in combination. (a,b) Western blot analysis was performed to elucidate the response of indicated signaling proteins in TS543 spheroids to CBD (10 μM) alone or in combination with LY294002 (40 μM), SB203580 (20 μM), Bay11-7082 (20 μM), BMS344551 (10 μM) 24 h after treatment. Original blots are shown in the “Supplementary information” section. After protein transfer, blot membranes were cut in two (or three) parts, which contained high molecular weight and low molecular weight proteins, respectively, and utilized for incubation with corresponding primary antibodies. (c,d) Combined treatment of TS543 spheroids using indicated combinations of CBD (5–20 μM), SP600125 (20 μM), LY294002 (40 μM) + BMS344551 (10 μM), Bay11-7082 (20 μM). AG490 (20 μM) and rapamycin (0.1 μM). Cell viability assay was performed for spheroid cultures 48–72 h after indicated treatments. The pooled results of four independent experiments are shown. Error bars represent means ± S.D. (p < 0.05, Student’s t-test). Stars and dual stars indicate significant differences in cell viability after specified treatments. (e) TS543 spheroids were treated, as indicated for 48 h, then labeled by incubation with CellTracker (green) for 30 min and further analyzed using fluorescent microscopy. Scale bars = 200 μm.
Continuous culturing of TS543 spheroids (with the addition of the fresh medium) results in establishing 3D organoids by integration of individual spheroids (Fig. 9). 3D TS543 organoids displayed a strong sensitivity to suppressive effects of CBD, especially, in combination with γ-irradiation (Fig. 9a,c). Interestingly, the effect of CQ on the induction of cell death was substantially less pronounced, than CBD in organoid culture. Furthermore, combined treatments by CBD+CQ or CBD+CQ+10 Gy demonstrated some protective effects of CQ, highlighting a probability of autophagy-liked death in conditions of strong deficiency of nutritional resources. In contrast, the addition of small molecule inhibitors of AKT/NF-κB in culture media dramatically decreased the viability of TS543 organoids (Fig. 9b,c).

Treatment of TS543 organoid cultures by CBD, CQ, γ-irradiation and small molecule inhibitors, alone or in combination. (a,b) TS543 organoids were grown in serum-free media with growth factors for 10 days (see “Methods” section). Then they were treated, as indicated, with further incubation for an additional 72 h. For confocal imaging, cells were stained by CellTracker (green) 30 min. (c) Cell viability assay was performed for non-stained organoids using the luminescent detection of ATP levels. Cell viability was normalized by cell numbers. Error bars represent means ± S.D. (p < 0.05, Student’s t-test. Stars and arrows indicate a significant difference in cell viability after specified treatments.
The higher sensitivity of 3D cultures of U87MG and TS543 to CBD, compared to 2D U87MG, is an important observation that needs a specific elucidation. It can be also useful to investigate if under 3D conditions hypoxia pathway is induced in response to CBD treatment (maybe due to local worse diffusion of oxygen). First, we re-determined apoptotic/necrotic induction in TS543 spheroids and 2D U87MD using Annexin-V-FITC and PI staining after treatment with increased doses of CBD (0–40 μM) in the presence and the absence of CQ (20 μM) (Fig. 10a,b) that confirmed a dose-dependent pro-apoptotic function CBD and preferential pro-necrotic acceleration of CQ. Western blot analysis of signaling proteins related to regulation of oxidative stress-hypoxia, such as transcription factors Arsenite Resistance Protein 2 (ARS2) and Nuclear factor erythroid 2-Related Factor 2 (NRF2) and their downstream target heme oxygenase-1 (HO-1), demonstrated dramatic differences in the expression levels of these proteins in 2D U87MF compared to TS543 spheroids (Fig. 10c), while several other proteins, such as NF-κB p65-P and HIF1-α, exhibited relatively small variation. Addition of an inhibitor of HO-1 enzymatic activity, ZnPP (10 μM), caused significant upregulation of CBD-induced apoptosis in 2D U87MG culture that achieved the levels of CBD-induced apoptosis in TS543 cells (Fig. 10d).

Distinct effects of CBD treatment on TS543 spheroids and U87MG adherent culture. (a,b,d) Annexin-V-FITC and PI staining for determination of the early apoptotic, late apoptotic and secondary-necrotic cells after treatment by CBD (0–40 μM) or CBD+CQ (20 μM) was followed by the flow cytometry. ZnPP (20 μM), an inhibitor of heme oxiginase-1 (HO-1) activity, was added 30 min before CBD. (c) Western blot analysis of indicated proteins from 3D TS543 and 2D U87MG cell cultures 4 h after indicated treatments. Original blots are shown in the “Supplementary information” section. After protein transfer, blot membranes were cut in two (or three) parts, which contained high molecular weight and low molecular weight proteins, respectively, and utilized for incubation with corresponding primary antibodies. The center lanes in western blots (which contain proteins after additional treatments non-used in this paper) were removed.
Discussion
Killing efficiency of cannabinoids (CBD, THC and their combination CBD+THC) against GBM in vitro and in animal experiments has been elucidated in numerous studies during the last 15 years12–14,47–50. Additional investigations also confirmed a cytotoxic role of cannabinoids for several other types of cancer12,30,51. A number of studies demonstrated the efficiency of combined treatments of cannabinoids together with γ-irradiation in both cell culture and in animal experiments17,18,28,31. CBD treatment can induce apoptotic death in GBM using both the intrinsic and extrinsic death pathways52 while γ-irradiation results in enhancing apoptotic and non-apoptotic cell death, especially in the presence of ATM inhibitor31,53. However, CBD and THC could be involved in the regulation of additional cell signaling networks including autophagy.
Numerous investigations were performed regarding a dual role of autophagy (either protection or sensitization) to cure cancer22,54,55. Dramatic stimulation of autophagy by THC56 or by novel inducers, such as loperamide57, could trigger autophagy-linked cell death in GBM. On the other hand, many studies demonstrated a protective role of autophagy for the control of viability of cancer cells, permitting upregulation of cancer cell death that was induced by γ-irradiation or TMZ by repression of the general lysosome function and autophagic flux using CQ58–60. The major aim of the present study was to elucidate a protective effect of CBD-induced autophagy in human GBM cell cultures and, consequently, to initiate a suppression of autophagic flux by CQ for enhancing CBD- or CBD/γ-irradiation induced death of cancer cells. In our experiments, we compared treatment opportunities for a standard 2D culture of GBM cells and 3D cultures of GBM spheroids and microtumors, which represent more precise a model of tumors and could be used more adequately for evaluation of cancer-killing efficiency of testing drugs61.
In both models, we evaluated effects of CBD and γ-irradiation, alone or in combination with CQ, an inhibitor of the lysosomal function that also caused suppression of autophagic flux22, on the regulation of cell survival in GBM cells. We monitored upregulation of BECLIN-1, a biomarker of the early stage of autophagy and used a ratio of LC3-II/LC3-I proteins, as a characteristic biomarker of late autophagy. CBD was confirmed as a powerful inducer of autophagy 6 h after treatment of U87MG GBM cells. In contrast, γ-irradiation alone or in combination with ATM inhibitor did not notably affect or only marginally affect autophagic flux at this time point. On the other hand, it was also possible to see upregulation of the LC3 puncta formation 6 h after CBD treatment of GBM cells. In contrast, γ-irradiation induced a modest upregulation of LC3 levels in the cytoplasm and a very strong increase of LC3 levels in the nuclei of GBM cells 6 h after treatment (see Figs. 1c and and2b).2b). It was previously recognized that LC3 is abundant in the nucleus, and the nuclear pool of LC3 represented the primary source for the cytoplasmic LC3 in the process of nucleocytoplasmic translocation (after deacetylation of LC3) for autophagy induction62.
An increase in autophagic flux (determined using an LC3-II/LC3-I ratio) was evident in U87MG GBM cells 24 h after γ-irradiation, especially in the presence of ATM inhibitor (see Fig. 1b). Numerous studies demonstrated upregulation of autophagic flux in treated cancer cells that could be involved in the regulation of both cell survival and death54,56,59. We utilized in our experiments 2D GBM cultures of adherent cells, such as U87MG, T98G, and U118MG, 3D cultures of U87MG spheroids and microtumors and 3D cultures of TS543 spheroids and organoids. We revealed substantial differences in 2D and 3D culture responses to CBD, CQ and CBD+CQ treatments: suppression of CBD-induced late autophagy by CQ resulted in pronounced anti-protective effects for 2D cultures of U87MG and T98G cells while these effects were relatively modest (or even neutral) for 3D spheroids cultures of U87MG and TS543. Finally, for TS543 organoids, CQ treatment did not provide an increase of CBD/γ-irradiation-induced cell death (Fig. 9). Correspondingly, 3D U87MG and TS543 were much more sensitive to CBD, compared to 2D U87MG. A special investigation of signaling proteins responding to CBD-induced oxidative stress highlighted a protective role of NRF2 – HO-1 signaling pathway (which is highly active in 2D U87MG) against oxidative stress (Fig. 10).
Transcriptional regulation of autophagy is based on activation and co-interaction of transcription factors operating together with epigenetic mechanisms to control expression of the autophagy-related genes23,63. CBD treatment of cancer cells induced or suppressed critical signaling pathways, targeted transcription factors and modulated the expression of numerous genes, including cytokine genes (IL-1β, IL-6 and IL-8) that could also regulate autophagy28,64–67. We further elucidated a role of distinct signaling pathways that could affect autophagy (among several other targets) in the regulation of cell death or survival. We confirmed CBD-mediated upregulation of JNK1/2-P and MAPK p38-P activities, which were involved in positive regulation of autophagy in GBM, and CBD-mediated suppression of AKT-mTOR pathway, which is known as the master inhibitor of autophagy. Finally, a stimulation of autophagy with the simultaneous repression of an inhibitor of autophagy resulted in substantial upregulation of autophagy22,27,28,30,31,56,68 in different cancers including proneural GB. On the other hand, JNK, MAPK p38, ERK1/2, and AKT-mTOR control numerous cell regulatory functions, including gene expression, protein synthesis and cell metabolism during cell survival and death. Furthermore, CBD treatment exhibited a varied and cell-specific degree of downregulation of NF-κB pathway that was highly active in GBM22. Our data demonstrated the high efficiency of combined treatment of CBD together with small molecule inhibitors of PI3K-AKT/NF-κB and IKK-NF-κB that effectively suppressed these pathways for upregulation of cell death levels in TS543 GBM spheroid cultures. Autophagy regulation could also be mediated by the JAK2-STAT3 pathway controlling expression of numerous biologically active targets69 and possessing anti-autophagic activity70. Indeed, CBD-activated the JAK2/STAT3 signaling pathway could induce BCL2 gene expression and via a feedback mechanism, using protein-protein interaction of BECLIN-1 and BCL2, to suppress autophagy66. Furthermore, cytoplasmic STAT3 could also be involved in suppression autophagy70. Our experiments with inhibition of JAK2/STAT3 pathway (utilizing AG490, a JAK2 inhibitor, and STAT3 inhibitor-6) demonstrated STAT3-dependent pro-surviving functions in CBD-targeted GBM cells (see Fig. 4f–h) and substantial change in the live-death balance at the CBD-induced conditions in the presence of STAT3 inhibitors.
The main observations and results of the present study are: i) substantial levels of basal autophagy in human GBM lines U87MG and T98G that were grown as 2D cultures; ii) protective effects of CBD-induced autophagy on survival of 2D U87MG and T98G cells that could be blocked by CQ; iii) a partial suppression of apoptosis but upregulation of total cell death levels by combination of CBD+CQ in these cells; iv) high sensitivity of 3D U87MG spheroids to CBD-induced death and less pronounced (or neutral) role of CQ co-treatment; v) high sensitivity of 3D TS543 spheroids to CBD-induced death while additional co-treatment with CQ demonstrated modest or neutral effects. Taken together, the results obtained highlighted a protective role of autophagy against CBD-induced glioma death in 2D cultures. On the other hand, suppression of autophagic flux plays a relatively minor role for an additional increase of high levels of CBD-induced death in 3D glioma cultures where NRF2-HO-1 signaling was relatively low effective. In contrast, a combination of CBD with small molecule inhibitors of cell signaling pathways (such as PI3K-AKT/NF-κB, IKK-NF-κB and JAK2-STAT3) potentially could be more effective for the final killing of 3D glioma cultures. Additional preclinical research using animal models could be important for the continuation of these studies.
Materials and Methods
Reagents
PI3K inhibitor LY294002, IKKβ inhibitor BMS345541 were obtained from Sigma (St. Louis, MO, USA), IKKα inhibitor Bay11-7082, MAPK p38 inhibitor SB203580, JNK1/2 inhibitor SP600125, JAK2 inhibitor AG490, STAT3 inhibitor-6 and ATM kinase inhibitor KU60019A were purchased from Calbiochem (La Jolla, CA, USA). Cannabidiol (exempt preparation; #90081) was obtained from Cayman Chemical (Ann Arbor, MI).
Human embryonic fibroblasts (TIG3) immortalized human fetal astrocytes (IHFA) in culture
TIG3 and IHFA were cultured in DMEM with 10% FBS, as previously described71.
2D and 3D GBM cell cultures
Three human GBM lines, which were used in the present study, were obtained from the ATCC: i) U87MG (HTB-14): very tumorigenic, highly invasive, highly rearranged hypodiploid with several hundred mutations, TP53wt, PTENmut; ii) U118MG (HTB-15): tumorigenic, moderate invasive; genome average copy number 2.2, several hundred mutations, TP53mut, PTENmut; iii) T98G (CRL-1690): non-tumorigenic; moderate invasive, highly rearranged hypopentaploid/hypohexaploid, several hundred mutations, TP53mut, PTENmut)72,73. These GBM lines were grown as 2D culture in DMEM with 5–10% FBS and 1% pyruvate74.
For neurosphere (spheroid) formation, U87MG GBM cells were cultured in the serum-free media DMEM/F12 supplemented with 2 mM GlutaMAX, bFGF (20 ng/ml), EGF (20 ng/ml) and B27 supplement (2%). All reagents were obtained from Gibco/Life Technologies (Carlsbad, CA, USA). After 10 passages, U87MG neurosphere culture was significantly enriched by CD133+ cells. Additionally, human TS543 proneural glioma cell line at low passage was grown as neurosphere culture75 using in vitro proliferation kit for human neural stem cells NeuroCult NS-A (StemCell Technology, #05751) supplemented with heparin (2 μg/ml), human bFGF (10 ng/ml) and human EGF (20 ng/ml). Prolonged culturing of TS543 of U87MG spheroids during 10 days resulted in the fusion of neurospheres into organoids.
Alternatively, 3D U87MG microtumors were obtained after culturing GBM cells in the matrices of Matrigel-collagen in serum-free media with growth factor supplements. For this purpose, collagen gel solution was prepared on ice by mixing together the following stock solutions: 0.35% collagen solution, medium, and 1 M Hepes (pH 7.4) in a ratio of 8:1:1 by volume. This solution was then mixed with Matrigel (BD Biosciences, Bedford, MA) in a ratio of 3:1 by volume and dropped into 12-well plates. The U87MG cell solution was injected as a bolus of cells in 1 μl aliquots (1000 cells) in several points of the matrices. The gel matrices were then overlaid with medium with supplements. Cells embedded in gel matrices were incubated at 37 °C in a humidified incubator (5% CO2, 95% air) for 1 h, treated with CBD, CQ, and γ-irradiation and further incubated for 1–6 days. The medium was refreshed every 24 h.
Immunocytochemistry analysis
Cells were fixed with 4% paraformaldehyde in PBS for 60 min. Immunochemical staining was performed using standard protocols. GBM cells were stained for LC3A/B (rabbit Ab #4108 from Cell Signaling) and α-Tubulin (mAb #T5168 from “Sigma”). The secondary Abs were Alexa Fluor 594 goat anti-mouse IgG and Alexa Fluor 488 goat anti-rabbit IgG from Molecular Probes/Life Technologies (Carlsbad, CA, USA). A Nikon A1 scanning confocal microscope on an Eclipse TiE microscope stand (Nikon Instruments, Melville, NY) was used for immunofluorescence image acquisition (as Z-series projection) and analysis. Images were further analyzed and quantified using ImageJ software (NIH).
Irradiation procedures
To determine sensitivity to X-rays, GBM cells and normal control cells were exposed to radiation from a Gammacell 40 137Cs irradiator (dose rate, 0.82 Gy/min) at Columbia University. Six to 72 h after irradiation, cells were stained with Annexin-V-FITC + PI and analyzed by flow cytometry for apoptosis studies. Alternatively, cells were labeled using CytoTracker and analyzed by fluorescent microscopy. Non-stained cells were also used for cell viability assay.
Cell viability and death studies
The CellTiter-Glo Luminescent Cell Viability Assay (“Promega”, Madison, WI53711, USA; #G7570) based on quantitation of ATP due to ATP-dependent luminescent signal, was performed using the manufactory’s protocol. For induction of death, cells were exposed to CBD (5–20 μM) and γ-irradiation (5–10 Gy) alone or in combination. In some experiments, small molecule inhibitors of cell signaling pathways were additionally used. For apoptosis and necrosis detection, staining of fresh cells by Annexin-V-FITC + Propidium Iodide (PI) and quantifying the percentage of Annexin-V-FITC-positive, PI-negative cells (the early apoptotic), Annexin-V-FITC-positive, PI-positive cells (the late apoptotic) and Annexin-V-FITC-negative, PI-positive cells (the secondary necrotic) was performed using reagents from BD Pharmingen (San Diego, CA) that was followed by the flow cytometry on FACSCANTO II flow cytometer (Becton Dickinson) using the FCS Express program. Cell cycle-apoptosis analysis was performed after fixation – permeabilization of cells with 70% ethanol, PI staining of DNA and flow cytometry.
Western blot analysis
Total cell lysates (40 μg protein) were resolved on SDS-PAGE, and processed according to standard protocols. Proteins were transferred onto a Immobilon PVDF (polyvinyldilene difluoride) membrane. Then membrane was cut in two parts, which contained high molecular weight and low molecular weight proteins, respectively, and utilized for incubation with corresponding primary antibodies. The antibodies used for Western blotting included anti-β-Actin mouse mAb #A5316 (Sigma, St. Louis, MO, USA). The antibodies to human antigens obtained from Cell Signaling (Danvers, MA) included phospho-p44/p42 MAPK (Erk1/2) (T202/Y204) rabbit mAb #4377; p44/p42 MAPK (Erk1/2) Ab #9102; phospho-SAPK/JNK (Thr183/Tyr185) rabbit mAb #4668; SAPK/JNK Ab #9252; phospho-cJUN (Ser73) Ab#9164; cJUN mouse mAb #2315; phospho-p38 MAPK (Thr180/Tyr182) rabbit mAb #4511; phospho-ATF2 (Thr71) Ab #9221; ATF2 rabbit mAb #9226; phospho-AKT1 (Ser473) #927; AKT1 Ab #9272; phospho-NF-κB p65 (Ser568) rabbit mAb #3033; NF-κB p65 rabbit mAb #4764; phospho-STAT3 (Tyr705) Ab #9131; phospho-STAT3 (Ser727) Ab #9134; STAT3 rabbit mAb #4904; phospho-p53 (Ser15) Ab #9284; p53 Ab #9282; phospho-beta-Catenin (Ser552) rabbit Ab #9566; beta-Catenin rabbit mAb #8480; phospho-mTOR (Ser2448) rabbit Ab #297; mTOR rabbit Ab #2972; Beclin-1 rabbit mAb #3495; LC3A/B Ab #4108; Caspase-9 (human specific) Ab #9502; Caspase-3 Ab #9662; HIF-1α mouse mAb #79233. The secondary antibodies were conjugated to horseradish peroxidase; signals were detected using the ECL system (Thermo Scientific, Rockford, IL, USA).
Suppression of cell signaling pathways by specific inhibitors
We performed specific inhibition of several signaling pathways using JNK1–3 inhibitor SP600125 (20 μM), MAPK p38 inhibitor SB203580 (20 μM), IKKβ-NF-κB inhibitor BMS345541 (10 μM); IKKα-NF-κB inhibitor Bay11-7082 (20 μM), PI3K-AKT inhibitor LY294002 (25–40 μM), JAK2-STAT3 inhibitor AG490 (20 μM) and STAT3 inhibitor-6 (40 μM). Western blotting with Abs to active forms of targeted proteins was used to evaluate the efficacy of inhibition. We assessed changes in cell death levels under these conditions. All inhibitors were dissolved in DMSO (0.1%) that was used as a control vehicle. CQ, an inhibitor of autophagic flux was dissolved in water and used at final concentration of 10–40 μM.
Statistical analyses of data
Data from four to five independent experiments were calculated as means and standard deviations. Comparisons of results between treated and control groups were made by the Students’ t-tests. A p-value of 0.05 or less between groups was considered significant.
- PMID: 32060308
- DOI: 10.1038/s41598-020-59468-4
-
Author Correction | Open Access | Published:
Vladimir N. Ivanov, Peter W. Grabham, Cheng-Chia Wu & Tom K. Hei
Scientific Reports volume 11, Article number: 18616 (2021)
Correction to: Scientific Reports https://doi.org/10.1038/s41598-020-59468-4, published online 14 February 2020
The original version of this Article contained errors in Figures 1 and 10, and in the Supplementary Information file. Figure 1a was assembled with two incorrect images of western blots for “ATM” and “p53-P(S15)”. Figure 1b was assembled with an incorrect image of western blot “LC3” at 24 h (left side), which contained an error in treatment condition and a transfer error for lane 1. The corresponding data for Figures 1 and 10 were also corrected in the Supplementary Information file. In addition, an error in treatment conditions for the western blots in Figure 10 was discovered. As a result, the western blots for “TS543”, “CBD (40)” in panel c were removed.
Figure 1 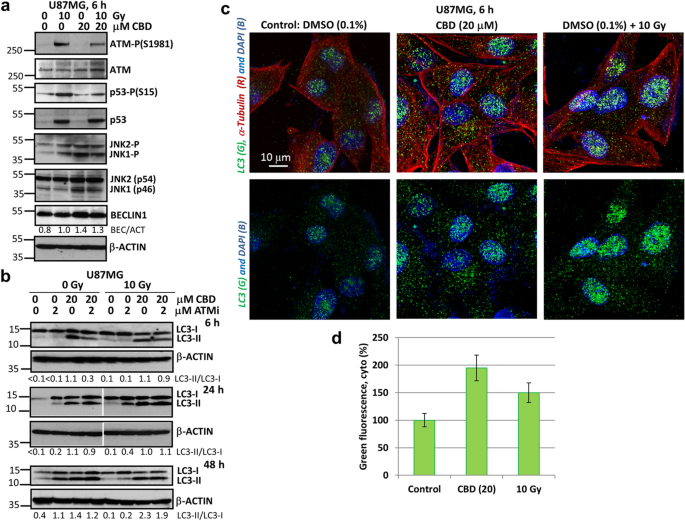
CBD induced autophagy in U87MG GBM 2D cell culture. (a) Western blot analysis of cell signaling proteins was performed 6 h after specified treatment of U87MG cells with CBD (20 μM) and γ-irradiation (10 Gy), alone or in combination. (b) Western blot analysis of LC3-II and LC3-I autophagy-related proteins was performed 6 h, 24 h and 48 h after treatments of GBM cells with CBD (20 μM), ATMi (2 μM) and γ-irradiation (10 Gy), alone or in combination. Original blots are shown in the “Supplementary information” section. After protein transfer, blot membranes were cut in two (or three) parts, which contained high molecular weight and low molecular weight proteins, respectively. The delineation of membranes was based on the well-known apparent molecular weight of investigated proteins. Cutting membranes were utilized for incubation with corresponding primary antibodies. The center lanes in LC3-I/II and β-ACTIN 24 h blots (which contain protein sample after an additional treatment non-used in this paper) were removed. (c) The LC3 puncta formation after indicated treatments of U87MG cells was detected using confocal microscopy with anti-LC3 Ab (green), anti-α-Tubulin Ab (red) and DAPI (blue). Images are shown with scale bar = 10 μm. (d) Relative levels of cytoplasmic green fluorescence (LC3) were determined using confocal images.
Figure 10 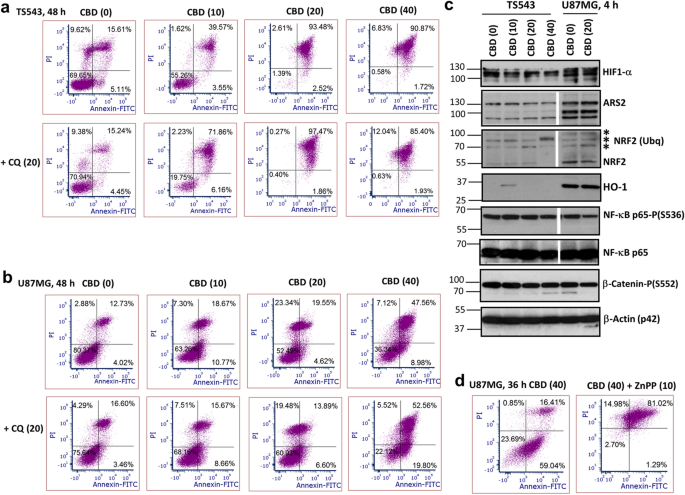
Distinct effects of CBD treatment on TS543 spheroids and U87MG adherent culture. (a,b,d) Annexin-V-FITC and PI staining for determination of the early apoptotic, late apoptotic and secondary-necrotic cells after treatment by CBD (0–40 μM) or CBD+CQ (20 μM) was followed by the flow cytometry. ZnPP (20 μM), an inhibitor of heme oxiginase-1 (HO-1) activity, was added 30 min before CBD. (c) Western blot analysis of indicated proteins from 3D TS543 and 2D U87MG cell cultures 4 h after indicated treatments. Original blots are shown in the “Supplementary information” section. After protein transfer, blot membranes were cut in two (or three) parts, which contained high molecular weight and low molecular weight proteins, respectively, and utilized for incubation with corresponding primary antibodies. Two center lanes in gel blots for ARS2, NRF2, NF-κB p65-P and NF-κB p65 (which contain proteins after additional treatments non-used in this paper) were removed. Stars indicate ubiquitinated NRF2.
Consequently, in the legend of Figure 10,“Two center lanes in gel blots for ARS2, NRF2, NF-κB p65-P and NF-κB p65 (which contain proteins after additional treatments non-used in this paper) were removed. Stars indicate ubiquitinated NRF2.”
now reads:
“The center lanes in western blots (which contain proteins after additional treatments non-used in this paper) were removed.”
The original Figures 1 and 10 and accompanying legends and the original Supplementary Information file are provided below. The original Article and accompanying Supplementary Information file have been corrected.
Author information


