

 Oncogene (2013) 32, 2534–2542 & 2013 Macmillan Publishers Limited All rights reserved 0950-9232/13
Oncogene (2013) 32, 2534–2542 & 2013 Macmillan Publishers Limited All rights reserved 0950-9232/13www.nature.com/onc
ORIGINAL ARTICLE
The orphan receptor GPR55 drives skin carcinogenesis
and is upregulated in human squamous cell carcinomas
E Pe ́rez-Go ́mez1, C Andradas1, JM Flores2, M Quintanilla3, JM Paramio4, M Guzma ́n1,5 and C Sa ́nchez1
G protein-coupled receptors (GPCRs) control crucial physiological processes and their dysfunction contributes to various human diseases, including cancer. The orphan GPCR GPR55 was identified and cloned more than a decade ago, but very little is known about its physio-pathological relevance. It has been recently shown that GPR55 controls the behavior of human cancer cell lines in culture and xenografts. However, the assessment of the actual role of this receptor in malignant transformation in vivo is hampered by the lack of studies on its functional impact in clinically-relevant models of cancer. Here we demonstrate that GPR55 drives mouse skin tumor development. Thus, GPR55-deficient mice were more resistant to DMBA/TPA-induced papilloma and carcinoma formation than their wild-type littermates. GPR55 exerted this pro-tumor effect primarily by conferring a proliferative advantage on cancer cells. In addition, GPR55 enhanced skin cancer cell anchorage-independent growth, invasiveness and tumorigenicity in vivo, suggesting that it promotes not only tumor development but also tumor aggressiveness. Finally, we observed that GPR55 is upregulated in human skin tumors and other human squamous cell carcinomas compared with the corresponding healthy tissues. Altogether, these findings reveal the pivotal importance of GPR55 in skin tumor development, and suggest that this receptor may be used as a new biomarker and therapeutic target in squamous cell carcinomas.
Oncogene (2013) 32, 2534–2542; doi:10.1038/onc.2012.278; published online 2 July 2012
Keywords: GPR55; G protein-coupled receptors; skin carcinogenesis; squamous cell carcinoma; cannabinoids
INTRODUCTION
G protein-coupled receptors (GPCRs) are the direct or indirect target of 450% of current therapeutic drugs. In the context of oncology, however, despite the increasing evidence showing a link between GPCR deregulation and cancer, just a few GPCRs are being exploited as targets of chemotherapeutic agents. Indirect lines of evidence suggest that the orphan GPCR GPR55 may have a role in cancer physiopathology. Thus, increased levels of the phospholipid lysophosphatidylinositol (LPI), a putative GPR55 endogenous ligand,1 have been found in plasma and ascites from patients with ovarian cancer compared with healthy women or with women with non-cancerous pathologies.2,3 In addition, epithelial cells4 and fibroblasts5 are able to generate mitogenic LPI upon ras-driven transformation. More recently, it has been shown that GPR55 modulates cancer cell migration6 and proliferation7,8 in vitro, and tumor growth in a xenograft-based model of glioblastoma.7 However, the actual role of GPR55 in malignant transformation in vivo remains unknown. One of the best established paradigms for studying the mechanisms underlying this process is the mouse skin model of two-stage carcinogenesis.9 Animals subjected to this experimental protocol evolve through different stages of cancer progression: first, in the ‘initiation’ phase, key genes are mutated in keratinocyte stem cells by topical exposure to the mutagenic agent 7,12- dimethylbenz[a]anthracene (DMBA).9 After the initiation stage, the population of mutated cells expands as a result of the repeated topical application of a proliferation-inducing agent such as the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA), UV radiation or wounding. During this ‘promotion’ stage, animals develop benign hyperplastic skin lesions termed papillomas.9 With variable frequency these structures may progress to malignant invasive squamous cell carcinomas or the more aggressive spindle cell carcinomas. This ‘progression’ stage occurs independently of tumor promoters and is the consequence of the genetic alterations accumulated during the proliferation of initiated cells.9 Here, by using this experimental protocol, as well as other in vivo and in vitro approaches, we demonstrate that GPR55 has an essential role in skin tumor development.
G protein-coupled receptors (GPCRs) are the direct or indirect target of 450% of current therapeutic drugs. In the context of oncology, however, despite the increasing evidence showing a link between GPCR deregulation and cancer, just a few GPCRs are being exploited as targets of chemotherapeutic agents. Indirect lines of evidence suggest that the orphan GPCR GPR55 may have a role in cancer physiopathology. Thus, increased levels of the phospholipid lysophosphatidylinositol (LPI), a putative GPR55 endogenous ligand,1 have been found in plasma and ascites from patients with ovarian cancer compared with healthy women or with women with non-cancerous pathologies.2,3 In addition, epithelial cells4 and fibroblasts5 are able to generate mitogenic LPI upon ras-driven transformation. More recently, it has been shown that GPR55 modulates cancer cell migration6 and proliferation7,8 in vitro, and tumor growth in a xenograft-based model of glioblastoma.7 However, the actual role of GPR55 in malignant transformation in vivo remains unknown. One of the best established paradigms for studying the mechanisms underlying this process is the mouse skin model of two-stage carcinogenesis.9 Animals subjected to this experimental protocol evolve through different stages of cancer progression: first, in the ‘initiation’ phase, key genes are mutated in keratinocyte stem cells by topical exposure to the mutagenic agent 7,12- dimethylbenz[a]anthracene (DMBA).9 After the initiation stage, the population of mutated cells expands as a result of the repeated topical application of a proliferation-inducing agent such as the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA), UV radiation or wounding. During this ‘promotion’ stage, animals develop benign hyperplastic skin lesions termed papillomas.9 With variable frequency these structures may progress to malignant invasive squamous cell carcinomas or the more aggressive spindle cell carcinomas. This ‘progression’ stage occurs independently of tumor promoters and is the consequence of the genetic alterations accumulated during the proliferation of initiated cells.9 Here, by using this experimental protocol, as well as other in vivo and in vitro approaches, we demonstrate that GPR55 has an essential role in skin tumor development.
RESULTS
GPR55 is expressed in mouse epithelia
First, we analyzed the expression of GPR55 in different mouse- stratified epithelia. As shown in Supplementary Figure 1, the receptor was moderately expressed in mouse skin, specifically in the epidermis (Supplementary Figure 1A). A similar pattern of GPR55 expression was found in other mouse-stratified epithelia. Thus, a discreet GPR55 immunoreactivity was detected in the epithelial compartments of the oral cavity (Supplementary Figure 1B), esophagus (Supplementary Figure 1C) and stomach (Supplementary Figure 1D). Interestingly, GPR55-positive cells were mainly located in the most proliferative layers of those epithelia (Supplementary Figure 1). Controls of the specificity of GPR55 staining are provided in Supplementary Figure 2.

GPR55 is expressed in mouse epithelia
First, we analyzed the expression of GPR55 in different mouse- stratified epithelia. As shown in Supplementary Figure 1, the receptor was moderately expressed in mouse skin, specifically in the epidermis (Supplementary Figure 1A). A similar pattern of GPR55 expression was found in other mouse-stratified epithelia. Thus, a discreet GPR55 immunoreactivity was detected in the epithelial compartments of the oral cavity (Supplementary Figure 1B), esophagus (Supplementary Figure 1C) and stomach (Supplementary Figure 1D). Interestingly, GPR55-positive cells were mainly located in the most proliferative layers of those epithelia (Supplementary Figure 1). Controls of the specificity of GPR55 staining are provided in Supplementary Figure 2.
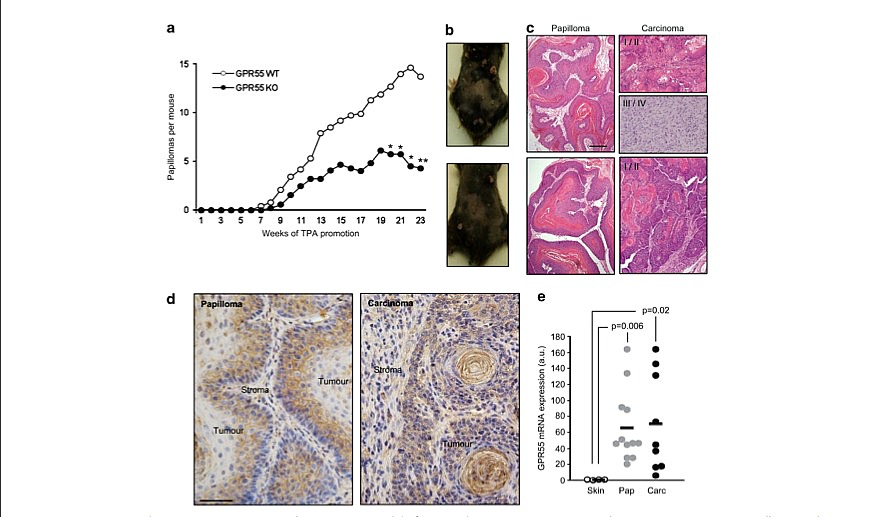
Figure 1. GPR55 drives tumor promotion in the two-stage model of mouse skin carcinogenesis. WT and GPR55 KO mice were topically treated with a single dose of DMBA followed by repeated applications of TPA for 22 weeks. Animals were then left untreated until week 38 after DMBA initiation to allow the development of carcinomas. (a) Tumor multiplicity, expressed as number of papillomas per mouse, at the indicated times. *Po0.05; **Po0.01 vs WT mice. Representative images of WT (upper panel) and GPR55 KO (lower panel) papilloma-bearing mice, (b) and papilloma and carcinoma architecture (H&E staining, c). (d) Immunohistochemical analysis of GPR55 in a representative WT mouse- derived papilloma (left panel) and carcinoma (right panel). GPR55 appears in brown and cell nuclei in blue. (e) GPR55 mRNA expression in normal skin, papillomas and carcinomas, as determined by RTQ–PCR. Results are expressed in arbitrary units. Scale bar in c, 500 mm; Scale bar in d, 100 mm.
GPR55 in skin tumor development
E Pe ́rez-Go ́mez et al
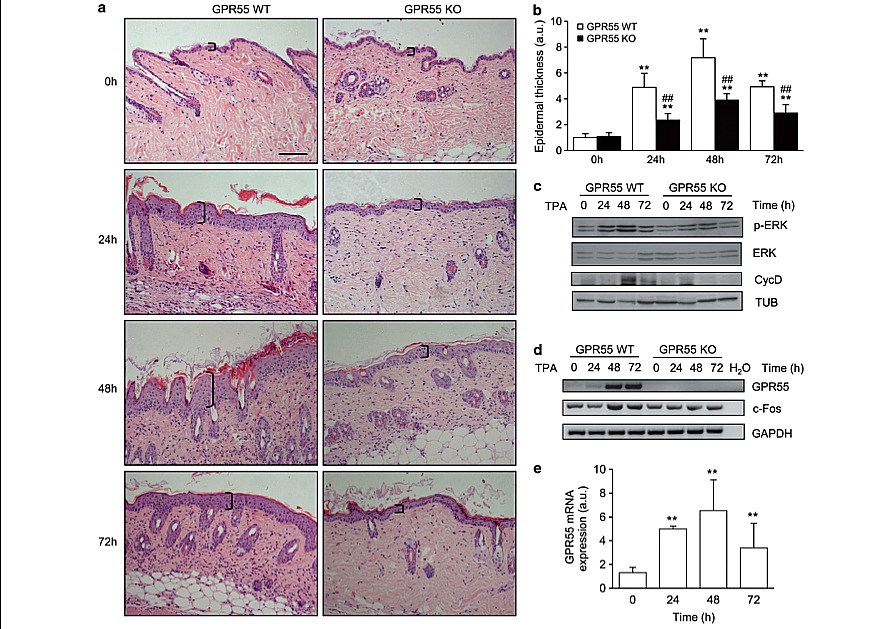
Figure 2. GPR55 mediates TPA-induced epidermal hyperproliferation. WT and GPR55 KO mice were subjected to a single skin topical application of TPA. The following features were analyzed at the indicated times: skin architecture (a) and epidermal thickness (b) by H&E staining; the levels of different markers of cell proliferation by western blot (c) and semi-quantitative PCR (d); and GPR55 levels in WT mouse skin by RTQ–PCR (e). **Po0.01 vs t0 (time of TPA application); ##Po0.01 vs the corresponding group of WT mice. Scale bar, 200 mm.
E Pe ́rez-Go ́mez et al
Figure 2. GPR55 mediates TPA-induced epidermal hyperproliferation. WT and GPR55 KO mice were subjected to a single skin topical application of TPA. The following features were analyzed at the indicated times: skin architecture (a) and epidermal thickness (b) by H&E staining; the levels of different markers of cell proliferation by western blot (c) and semi-quantitative PCR (d); and GPR55 levels in WT mouse skin by RTQ–PCR (e). **Po0.01 vs t0 (time of TPA application); ##Po0.01 vs the corresponding group of WT mice. Scale bar, 200 mm.
GPR55-deficient mice are resistant to skin tumor development Next, we analyzed the role of GPR55 in skin carcinogenesis. Skin tumors were induced in wild-type (WT) and GPR55-deficient mice by topical treatment with a single dose of DMBA followed by a repeated administration of TPA for 22 weeks. GPR55-deficient mice are viable and show no major differences compared with WT littermates, aside from a slightly altered bone architecture10 and a decreased mechanical hyperalgesia in response to inflammatory and neuropathic pain.11 After the DMBA/TPA challenge, we observed no differences between WT and GPR55-deficient mice either in the latency (time from treatment initiation to tumor development), the size (data not shown) or in the histopathological features of the papillomas (Figure 1c, left panels). However, GPR55 KO mice developed significantly less papillomas per animal than their WT littermates (Figures 1a and b). In addition, the frequency of malignant conversion, as determined by the percentage of papillomas that progressed into carcinomas, was significantly lower in the GPR55 KO population than in the WT population (Table 1). Moreover, whereas all the carcinomas in the GPR55 KO group were well differentiated (grades I and II), 22% of the WT carcinomas were poorly differentiated (classified as grade III–IV, that is, highly aggressive) (data not shown). Of interest, the expression of GPR55 in the skin lesions of WT animals was high in the tumor compartment and remained low in the non-tumor tissue (Figure 1d). Likewise, GPR55 mRNA levels were significantly higher in papillomas and carcinomas as compared with normal mouse skin (Figure 1e).
GPR55 deficiency reduces TPA-induced epidermal hyperproliferation and inflammation in vivo
Exacerbated proliferation of initiated cells is crucial in the first stages of tumor development. Consequently, we analyzed whether GPR55 expression confers a proliferative advantage on epidermal cells in vivo in response to a promoting agent. We treated the skin of GPR55-deficient mice and their corresponding WT littermates topically with a single dose of TPA. No significant differences were found in the skin architecture of WT and GPR55 KO mice before TPA treatment (Figure 2a, upper panels). However, TPA challenge induced a remarkable increase in epidermal thickness in WT animals that was significantly attenuated in GPR55 KO mice (Figures 2a and b). Known drivers of cell proliferation in this model (phosphorylated ERK, c-Fos and Cyclin D1) were increased upon TPA treatment in WT mice, an effect that was not observed in the corresponding GPR55 KO animals (Figures 2c and d). Interestingly, TPA-induced hyperplasia in the WT population was accompanied by a strong upregulation of GPR55 levels in the skin (Figures 2d and e and Supplementary Figure 3). Proliferating cell nuclear antigen staining revealed that, after TPA challenge, the skin of GPR55-deficient mice presented significantly less proliferating cells than the skin of WT animals (Figure 3).
Exacerbated proliferation of initiated cells is crucial in the first stages of tumor development. Consequently, we analyzed whether GPR55 expression confers a proliferative advantage on epidermal cells in vivo in response to a promoting agent. We treated the skin of GPR55-deficient mice and their corresponding WT littermates topically with a single dose of TPA. No significant differences were found in the skin architecture of WT and GPR55 KO mice before TPA treatment (Figure 2a, upper panels). However, TPA challenge induced a remarkable increase in epidermal thickness in WT animals that was significantly attenuated in GPR55 KO mice (Figures 2a and b). Known drivers of cell proliferation in this model (phosphorylated ERK, c-Fos and Cyclin D1) were increased upon TPA treatment in WT mice, an effect that was not observed in the corresponding GPR55 KO animals (Figures 2c and d). Interestingly, TPA-induced hyperplasia in the WT population was accompanied by a strong upregulation of GPR55 levels in the skin (Figures 2d and e and Supplementary Figure 3). Proliferating cell nuclear antigen staining revealed that, after TPA challenge, the skin of GPR55-deficient mice presented significantly less proliferating cells than the skin of WT animals (Figure 3).
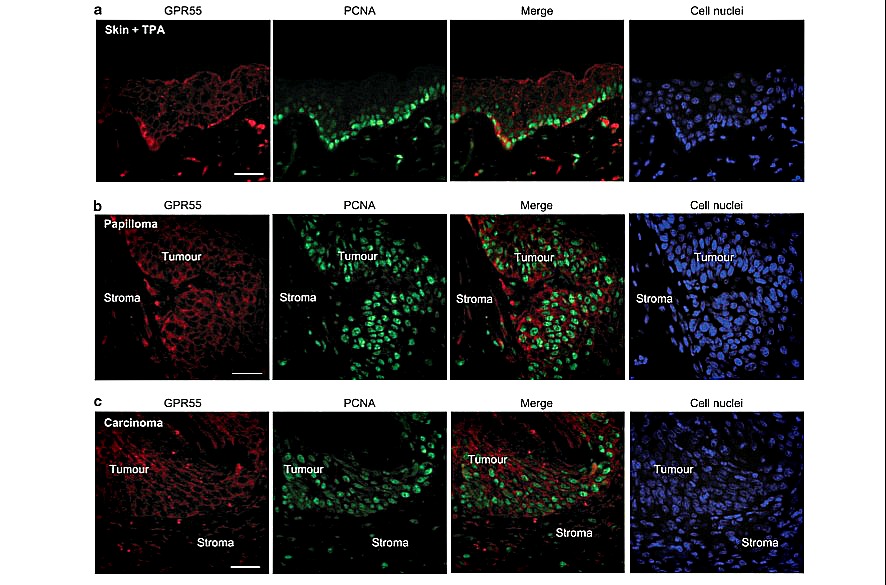
Moreover, simultaneous immunofluorescence analysis of GPR55 and proliferating cell nuclear antigen in these samples revealed that GPR55-expressing cells were those undergoing proliferation (Figure 4a). A similar colocalization between GPR55 expression and cell proliferation was observed in the mouse papillomas (Figure 4b) and carcinomas (Figure 4c) generated after DMBA/TPA treatment. Thus, GPR55 was present in the proliferative tumor compartment and virtually absent from the non-tumor and poorly proliferative stromal tissue (Figures 4b and c).
It is well described that, together with epidermal hyperproli- feration, TPA topical application on mouse skin induces local inflammation,11 which can be detected by the increase in dermal celullarity (Figure 2a). In agreement with this notion, we observed an increased number of inflammatory (CD45-positive) cells infiltrating the dermis (Supplementary Figures 4A and B), and augmented levels of ILb1 and TNFa (Supplementary Figures 4C and D). Importantly, we did not observe these characteristics in GPR55 KO mice (Figure 2 and Supplementary Figure 4).
It is well described that, together with epidermal hyperproli- feration, TPA topical application on mouse skin induces local inflammation,11 which can be detected by the increase in dermal celullarity (Figure 2a). In agreement with this notion, we observed an increased number of inflammatory (CD45-positive) cells infiltrating the dermis (Supplementary Figures 4A and B), and augmented levels of ILb1 and TNFa (Supplementary Figures 4C and D). Importantly, we did not observe these characteristics in GPR55 KO mice (Figure 2 and Supplementary Figure 4).
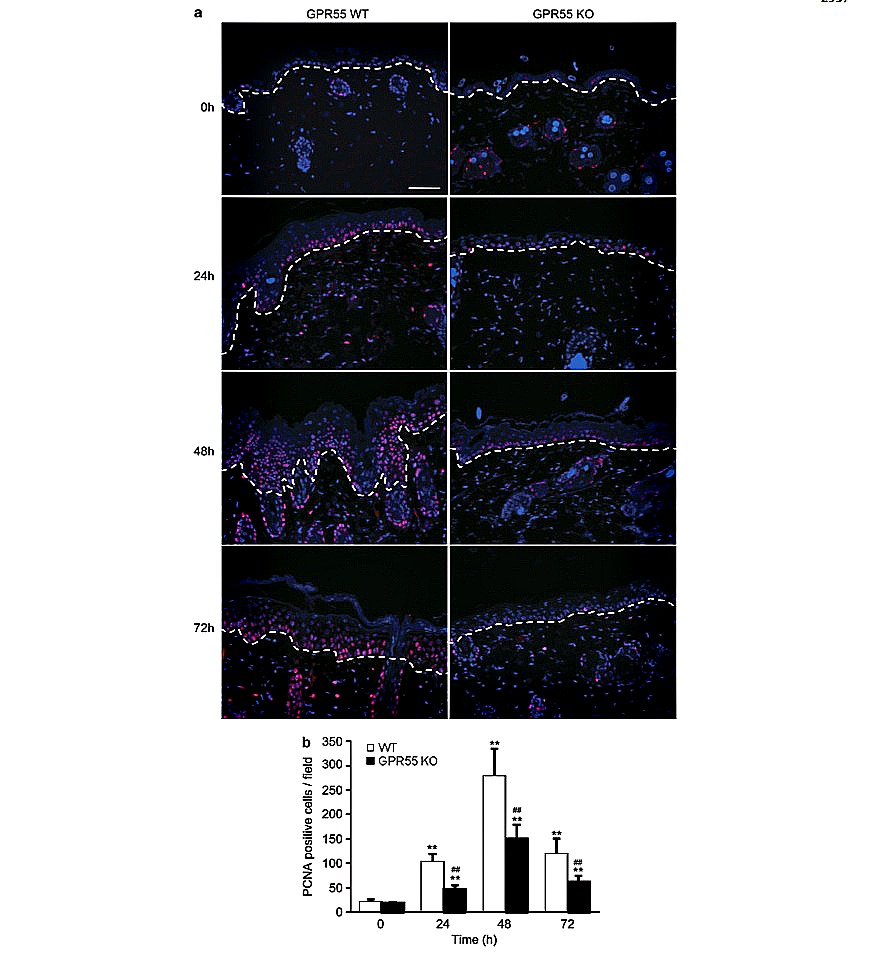
Figure 3. GPR55-deficient mice present reduced epidermal proliferation in response to TPA. (a) Immunofluorescence analysis of proliferating (that is, proliferating cell nuclear antigen (PCNA)-positive) cells in the skin of WT and GPR55 KO mice treated topically with a single TPA application. PCNA is stained in pink and cell nuclei in blue. (b) Quantification of PCNA-positive cells. **Po0.01 vs t0 (time of TPA application); ##Po0.01 vs the corresponding group of WT mice. Scale bar, 200 mm.
GPR55 in skin tumor development
E Pe ́rez-Go ́mez et al
Figure 4. Cells undergoing proliferation express GPR55. (a) WT mice were topically treated with a single dose of TPA for 48 h, and GPR55 (red) and proliferating cell nuclear antigen (PCNA, green) were analyzed by immunofluorescence. (b and c) GPR55 and PCNA protein expression was studied in a representative papilloma (b) and a representative carcinoma (c), generated in a WT mouse topically treated with a single dose of DMBA followed by the repeated application of TPA for 22 weeks. Cell nuclei are stained in blue. Scale bars, 100 mm.
E Pe ́rez-Go ́mez et al
Figure 4. Cells undergoing proliferation express GPR55. (a) WT mice were topically treated with a single dose of TPA for 48 h, and GPR55 (red) and proliferating cell nuclear antigen (PCNA, green) were analyzed by immunofluorescence. (b and c) GPR55 and PCNA protein expression was studied in a representative papilloma (b) and a representative carcinoma (c), generated in a WT mouse topically treated with a single dose of DMBA followed by the repeated application of TPA for 22 weeks. Cell nuclei are stained in blue. Scale bars, 100 mm.
Altogether, these observations support that GPR55 is an endogenous promoter of skin proliferation and inflammation.
GPR55 enhances the tumorigenic properties of skin cancer cells
To further demonstrate the functional relevance of GPR55 in skin tumorigenesis, we performed a series of in vitro and in vivo experiments with PDV skin carcinoma cells,12 which express GPR55 (Figure 5a). GPR55 expression was stably knocked down in these cells by means of selective shRNAs (Figure 5a). First, we observed that receptor silencing decreased the number of viable cells (Figure 5b). Additionally, anchorage-independent growth was impaired by GPR55 knockdown. Thus, GPR55-silenced cells generated significantly less and smaller colonies in soft agar than the corresponding control shRNA-transfected cells (Figure 5c). Invasion through Matrigel was also hampered in GPR55-deficient cells (Figure 5d). To further demonstrate the pro-oncogenic role of GPR55, we generated tumor xenografts by subcutaneous injection of shControl-PDV cells and shGPR55-PDV cells. We observed no differences in tumor incidence (100% in all cases) or latency, but receptor knockdown dramatically decreased tumor growth (Figure 5e). Collectively, these results demonstrate that GPR55 enhances the aggressiveness of skin cancer cells.
GPR55 enhances the tumorigenic properties of skin cancer cells
To further demonstrate the functional relevance of GPR55 in skin tumorigenesis, we performed a series of in vitro and in vivo experiments with PDV skin carcinoma cells,12 which express GPR55 (Figure 5a). GPR55 expression was stably knocked down in these cells by means of selective shRNAs (Figure 5a). First, we observed that receptor silencing decreased the number of viable cells (Figure 5b). Additionally, anchorage-independent growth was impaired by GPR55 knockdown. Thus, GPR55-silenced cells generated significantly less and smaller colonies in soft agar than the corresponding control shRNA-transfected cells (Figure 5c). Invasion through Matrigel was also hampered in GPR55-deficient cells (Figure 5d). To further demonstrate the pro-oncogenic role of GPR55, we generated tumor xenografts by subcutaneous injection of shControl-PDV cells and shGPR55-PDV cells. We observed no differences in tumor incidence (100% in all cases) or latency, but receptor knockdown dramatically decreased tumor growth (Figure 5e). Collectively, these results demonstrate that GPR55 enhances the aggressiveness of skin cancer cells.
GPR55 is overexpressed in human skin tumors and other human squamous cell carcinomas
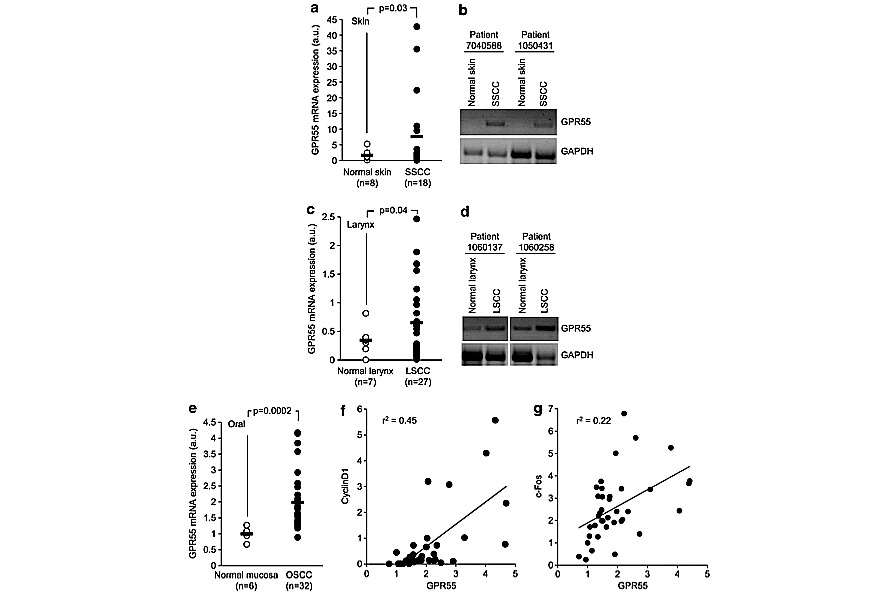
Finally, we analyzed GPR55 expression in human samples. GPR55 mRNA levels were hardly detectable in the normal skin (Figures 6a and b), but, interestingly, they were significantly increased in skin carcinomas compared with healthy tissue (Figures 6a and b). Similar results were found in other human squamous cell carcinomas such as larynx and oral cavity (Figures 6c–e) carcinomas, in which cancer tissue expressed significantly higher GPR55 mRNA levels than the corresponding non-tumor tissue. Oral carcinoma data were obtained from a previously published microarray dataset,13 and in this case Cyclin D1 and c-Fos tumor expression were also available. Of interest, tumor GPR55 expression positively correlated with tumor Cyclin D1 (Figure 6f) and c-Fos expression (Figure 6g), further supporting the link between GPR55 and cancer cell proliferation.
Finally, we analyzed GPR55 expression in human samples. GPR55 mRNA levels were hardly detectable in the normal skin (Figures 6a and b), but, interestingly, they were significantly increased in skin carcinomas compared with healthy tissue (Figures 6a and b). Similar results were found in other human squamous cell carcinomas such as larynx and oral cavity (Figures 6c–e) carcinomas, in which cancer tissue expressed significantly higher GPR55 mRNA levels than the corresponding non-tumor tissue. Oral carcinoma data were obtained from a previously published microarray dataset,13 and in this case Cyclin D1 and c-Fos tumor expression were also available. Of interest, tumor GPR55 expression positively correlated with tumor Cyclin D1 (Figure 6f) and c-Fos expression (Figure 6g), further supporting the link between GPR55 and cancer cell proliferation.
DISCUSSION
The orphan receptor GPR55 was identified and cloned in 1999.14 Experiments performed with GPR55-deficient mice have demonstrated that it participates in the control of inflammatory and neuropathic pain,11 and bone metabolism,10 but the broad distribution of this receptor throughout the body suggests that it may modulate many more physio-pathological functions.15 Regarding cancer, recent data suggest that GPR55 may be part of the molecular circuitry controlling tumor growth. Thus, this receptor promotes glioma, breast,7 prostate and ovarian8 cancer cell proliferation, and breast cancer cell migration6 in culture. It also induces tumor growth in a xenograft-based model of glioma.7 Here, by using a well-established model of chemically-induced carcinogenesis, we demonstrate that GPR55 has a prominent role in malignant growth. Specifically, we show that GPR55 deficiency makes mice significantly resistant to two-stage skin carcinogenesis. The main changes observed between WT and GPR55 KO mice concerned tumor multiplicity and not tumor incidence or tumor latency. We have demonstrated a link between GPR55 expression and increased cell proliferation. Thus, TPA- induced epidermal hyperproliferation in vivo is accompanied by an increase in GPR55 levels and, most important, is not evident in GPR55-deficient mice. Additionally, the cells undergoing proliferation in response to TPA are those with increased GPR55 expression. Moreover, when GPR55 is knocked down in skin cancer cells, their growth ability is hampered. Both orphan and well-characterized GPCRs have been previously associated with different aspects of cancer development, including aberrant cancer cell proliferation.16,17 Prominent examples of such GPCRs are lysophosphatidic acid (LPA) receptors. LPA is a lysophos- pholipid with potent mitogenic activity that produces its biological effects by binding to six specific GPCRs, namely LPA1-6.18,19 LPA signaling is frequently overactivated in human tumors and it is currently accepted to contribute to cancer progression by stimulating, among others, cancer cell proliferation.16,18 The similarities between LPA receptors and GPR55 are abundant. First, they are two of the few GPCRs engaged by lipid ligands. Although the pharmacology of GPR55 is still quite controversial, several reports have consistently demonstrated that GPR55 can be activated by LPI.1,10,20–25 Interestingly, LPI, as LPA, has mitogenic properties. Specifically, it has been described that epithelial cells4 and fibroblasts5 generate proliferation-inducing LPI upon ras-driven transformation. Cannabinoids, the active components of marijuana and their derivatives, which also have lipid nature, seem to activate this receptor as well, but in this case the evidence is not as consistent as for LPI: some cannabinoids were active in some reports and inactive in others, some were agonists in some studies and antagonists in others, and so on.26 Another interesting similarity between LPA receptors and GPR55 is that their respective ligands are upregulated in cancer. Thus, LPA is elevated in ascites27 and plasma28 of ovarian cancer patients and in the plasma of hepatocellular carcinoma patients.29 Regarding GPR55 ligands, LPI levels are increased in ascites3 and plasma2 from patients with ovarian cancer, and the endo- genous ligands of cannabinoid receptors (endocannabinoids) are enhanced in tumor vs non-tumor tissue in brain,30,31 prostate, endometrial32 and colorectal33 cancer patients. Together with their ligands, both LPA and GPR55 receptors themselves are frequently upregulated in neoplasias. LPA receptors, LPA2 in particular, are aberrantly expressed in a wide variety of human tumors, including thyroid,34 ovarian,35 colorectal,36 breast,37 colon, lung, uterus, rectum, testis,38 gastric39 and prostate40 carcinomas. The same is true for GPR55: herein, we present data showing that GPR55 is elevated in human carcinomas of the skin, larynx and oral cavity compared with the corresponding non-tumor tissues. These observations are in line with previous reports demonstrating that GPR55 mRNA is present in a wide collection of human cancer cell lines from breast,6,7 brain, cervix, skin, pancreas, liver,7 ovary, prostate,8 bile ducts41 and hematological origin.7,23,42 Moreover, the levels of this receptor are upregulated in breast, brain and pancreatic cancer compared with the corresponding healthy tissue, and correlate with tumor aggressiveness, that is, higher histological grades and, in the case of gliomas, lower survival rates.7
The orphan receptor GPR55 was identified and cloned in 1999.14 Experiments performed with GPR55-deficient mice have demonstrated that it participates in the control of inflammatory and neuropathic pain,11 and bone metabolism,10 but the broad distribution of this receptor throughout the body suggests that it may modulate many more physio-pathological functions.15 Regarding cancer, recent data suggest that GPR55 may be part of the molecular circuitry controlling tumor growth. Thus, this receptor promotes glioma, breast,7 prostate and ovarian8 cancer cell proliferation, and breast cancer cell migration6 in culture. It also induces tumor growth in a xenograft-based model of glioma.7 Here, by using a well-established model of chemically-induced carcinogenesis, we demonstrate that GPR55 has a prominent role in malignant growth. Specifically, we show that GPR55 deficiency makes mice significantly resistant to two-stage skin carcinogenesis. The main changes observed between WT and GPR55 KO mice concerned tumor multiplicity and not tumor incidence or tumor latency. We have demonstrated a link between GPR55 expression and increased cell proliferation. Thus, TPA- induced epidermal hyperproliferation in vivo is accompanied by an increase in GPR55 levels and, most important, is not evident in GPR55-deficient mice. Additionally, the cells undergoing proliferation in response to TPA are those with increased GPR55 expression. Moreover, when GPR55 is knocked down in skin cancer cells, their growth ability is hampered. Both orphan and well-characterized GPCRs have been previously associated with different aspects of cancer development, including aberrant cancer cell proliferation.16,17 Prominent examples of such GPCRs are lysophosphatidic acid (LPA) receptors. LPA is a lysophos- pholipid with potent mitogenic activity that produces its biological effects by binding to six specific GPCRs, namely LPA1-6.18,19 LPA signaling is frequently overactivated in human tumors and it is currently accepted to contribute to cancer progression by stimulating, among others, cancer cell proliferation.16,18 The similarities between LPA receptors and GPR55 are abundant. First, they are two of the few GPCRs engaged by lipid ligands. Although the pharmacology of GPR55 is still quite controversial, several reports have consistently demonstrated that GPR55 can be activated by LPI.1,10,20–25 Interestingly, LPI, as LPA, has mitogenic properties. Specifically, it has been described that epithelial cells4 and fibroblasts5 generate proliferation-inducing LPI upon ras-driven transformation. Cannabinoids, the active components of marijuana and their derivatives, which also have lipid nature, seem to activate this receptor as well, but in this case the evidence is not as consistent as for LPI: some cannabinoids were active in some reports and inactive in others, some were agonists in some studies and antagonists in others, and so on.26 Another interesting similarity between LPA receptors and GPR55 is that their respective ligands are upregulated in cancer. Thus, LPA is elevated in ascites27 and plasma28 of ovarian cancer patients and in the plasma of hepatocellular carcinoma patients.29 Regarding GPR55 ligands, LPI levels are increased in ascites3 and plasma2 from patients with ovarian cancer, and the endo- genous ligands of cannabinoid receptors (endocannabinoids) are enhanced in tumor vs non-tumor tissue in brain,30,31 prostate, endometrial32 and colorectal33 cancer patients. Together with their ligands, both LPA and GPR55 receptors themselves are frequently upregulated in neoplasias. LPA receptors, LPA2 in particular, are aberrantly expressed in a wide variety of human tumors, including thyroid,34 ovarian,35 colorectal,36 breast,37 colon, lung, uterus, rectum, testis,38 gastric39 and prostate40 carcinomas. The same is true for GPR55: herein, we present data showing that GPR55 is elevated in human carcinomas of the skin, larynx and oral cavity compared with the corresponding non-tumor tissues. These observations are in line with previous reports demonstrating that GPR55 mRNA is present in a wide collection of human cancer cell lines from breast,6,7 brain, cervix, skin, pancreas, liver,7 ovary, prostate,8 bile ducts41 and hematological origin.7,23,42 Moreover, the levels of this receptor are upregulated in breast, brain and pancreatic cancer compared with the corresponding healthy tissue, and correlate with tumor aggressiveness, that is, higher histological grades and, in the case of gliomas, lower survival rates.7
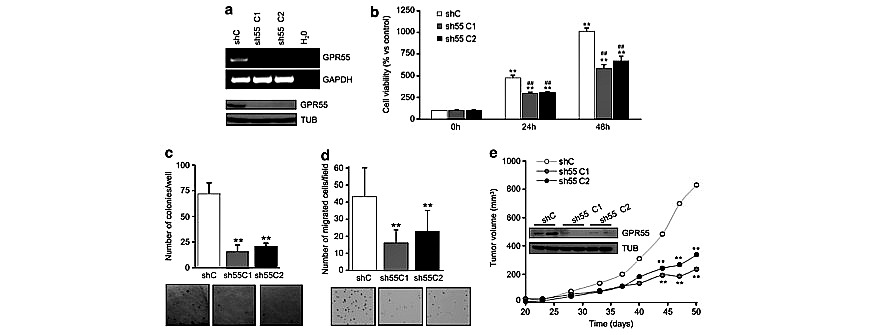
Figure 5. GPR55 enhances the pro-tumorigenic properties of epidermal cancer cells. GPR55 expression was stably knocked down in transformed PDV mouse keratinocytes by means of selective shRNAs. Two different clones of GPR55-silenced cells, sh55 C1 and sh55 C2, were used for the experiments. PDV cells stably transfected with a non-targeted shRNA (shC) were used as control. (a) Analysis of GPR55 levels by semi-quantitative PCR (upper panel) and western blot (lower panel). (b) Number of viable cells, as determined by the MTT-colorimetric test, at the indicated times. Results are expressed as % of shC cells at t0, set at 100%. **Po0.01 vs t0; ##Po0.01 vs the corresponding set of shC cells. Anchorage-independent growth (c), expressed as number of colonies per well formed in soft agar, and invasion capability toward FBS (d), expressed as number of migrated cells per field, of PDV cells. Representative images of the colonies (c) and the migrated cells (d) are shown in the lower panels. **Po0.01 vs shC cells. (e) PDV-derived subcutaneous xenograft growth. **Po0.01 vs shC tumors. Inset, GPR55 expression levels in two representative tumors generated by each cell line, as determined by western blot analysis.
GPR55 in skin tumor development
E Pe ́rez-Go ́mez et al
Figure 6. Expression of GPR55 in human samples. GPR55 mRNA levels were determined in a collection of skin- (a and b) and larynx-related (c and d) human samples. (a and c), GPR55 mRNA levels as determined by RTQ–PCR. (b and d) Representative semi-quantitative PCRs corresponding to pairs of tumor and non-tumor samples from the same patients. GPR55, c-Fos and Cyclin D1 mRNA expression data in oral cavity (e–g) were obtained from the microarray data set published in GEO database with the accession number GSE10121. (e) GPR55 mRNA levels in non-tumor and tumor samples. f and g represent GPR55 expression vs cyclin D1 and c-Fos expression, respectively. Results are expressed in arbitrary units. r2 1⁄4 coefficient of determination (Pearson correlation coefficient2). SSCC, skin squamous cell carcinoma; LSCC, larynx squamous cell carcinoma; OSCC, oral squamous cell carcinoma.
E Pe ́rez-Go ́mez et al
Figure 6. Expression of GPR55 in human samples. GPR55 mRNA levels were determined in a collection of skin- (a and b) and larynx-related (c and d) human samples. (a and c), GPR55 mRNA levels as determined by RTQ–PCR. (b and d) Representative semi-quantitative PCRs corresponding to pairs of tumor and non-tumor samples from the same patients. GPR55, c-Fos and Cyclin D1 mRNA expression data in oral cavity (e–g) were obtained from the microarray data set published in GEO database with the accession number GSE10121. (e) GPR55 mRNA levels in non-tumor and tumor samples. f and g represent GPR55 expression vs cyclin D1 and c-Fos expression, respectively. Results are expressed in arbitrary units. r2 1⁄4 coefficient of determination (Pearson correlation coefficient2). SSCC, skin squamous cell carcinoma; LSCC, larynx squamous cell carcinoma; OSCC, oral squamous cell carcinoma.
The cancer-related effects of LPA receptors and GPR55 are not restricted to the promotion of cancer cell proliferation. It has been described that LPA receptors drive cancer cell survival, migration, invasion and angiogenesis in in vitro and in vivo paradigms.43 Our findings suggest that GPR55 may promote other pro-tumorigenic actions aside from the induction of cancer cell proliferation. Thus, GPR55 knockdown impairs skin cancer cell anchorage- independent growth and migration, pointing to a potential role of the receptor in tumor development and aggresiveness. Previous data point in the same direction: (1) downregulation of GPR55 in prostate cancer cells decrease their ability to form colonies in soft agar;8 and (2) GPR55 ectopic expression increases migration of the poorly metastatic, low GPR55-expressing MCF-7 breast cancer cell line.6 Further studies are nonetheless required to determine whether GPR55 is definitely involved in cancer metastasis. We have also observed that GPR55 is necessary for TPA-induced dermal inflammation. Similarly, it has been shown that GPR55 KO mice are more resistant to chemically-induced colitis44 and that their basal and FCA-induced pattern of anti-inflammatory cytokines is different from that of their WT littermates.11 Together, these data suggest that GPR55 participates in the control of inflammatory responses, which may be of importance in tumor progression.
Collectively, our results strongly support that GPR55 has a pivotal role in skin tumor development, and suggest that this receptor, as part of the lysophospholipid signaling rheostat that is deregulated in cancer, may be used as a new therapeutic target and potential biomarker in skin cancer management.
Oncogene (2013) 2534 – 2542
MATERIALS AND METHODS
Animals and treatments
All procedures involving animals were performed with the approval of the Complutense University Animal Experimentation Committee in compliance with the European official regulations. GPR55 KO mice (B6;129S- Gpr55tm1Lex/Mmnc) were generated by Lexicon Pharmaceuticals (The Woodlands, TX, USA) and were obtained through the Mutant Mouse
Regional Resource Centers. This strain has a 129/SvEvBrd x C57BL6/J-mixed genetic background. As pure C57BL6/J mice are refractory to DMBA/ TPA-induced carcinogenesis,9 GPR55 KO animals were bred to the sensitive 129/Ola mice. Heterozygous animals from these progeny were intercrossed to generate the WT and GPR55 KO littermates used in this study. For the two-stage carcinogenesis experiments, tumors were induced on the shaved dorsal skin of WT (n 1⁄4 11) and GPR55 KO (n 1⁄4 11) mice by topical application of a single dose of DMBA (32 mg in 200 ml of acetone), followed by twice-weekly applications of TPA (12.5mg in 200ml of acetone) for 22 weeks. The number of tumors (42mm in diameter) was recorded weekly. TPA treatment was ended after 22 weeks, and animals were left untreated until week 38 after DMBA initiation to allow the development of carcinomas. Tumors were collected at different time points and histologically typed as previously described.12 For the acute TPA experiments, a single dose of TPA (12.5 mg) or the corresponding vehicle (200 ml of acetone) was applied onto the shaved dorsal skin of WT (n 1⁄4 10) and GPR55 KO (n 1⁄4 10) littermates. At the end of the experiments, mice were killed and tumors or skin were collected. Samples were divided into portions that were (1) frozen in Tissue-Tek (Sakura Finetek Europe, Zoeterwoude, The Netherlands) for immunofluorescence staining, (2) fixed in formalin for immunofluorescence, immunohistochemistry and H&E staining, or (3) snap-frozen for protein and RNA extraction. Samples were stored at -80 degrees C until analysis (except formalin-fixed samples, which were kept at room temperature.
Cell cultures, plasmids and transfections
The origin and culture conditions of the PDV-transformed murine keratinocytes has been described elsewhere.12 To stably knockdown GPR55, double-stranded oligonucleotides encoding shRNA against mouse GPR55 were inserted into the psi-U6-EGFP vector (GeneCopoeia, Rockville, MD, USA). A psi-U6-EGFP-unrelated sequence vector was used as control. Transfections were carried out using Lipofectamine-plus (Invitrogen, Barcelona, Spain) according to the manufacturer’s instructions. Cells were selected with puromycin (Sigma, St Louis, MO, USA), and individual clones were isolated using a FACS Vantage sorter (Becton Dickinson, Madrid, Spain). Cell viability was determined in 10% FBS (fetal bovine serum)- containing cell cultures by the MTT test (Sigma) according to manufacturer’s instructions.
Anchorage-independent growth
Oncogene (2013) 2534 – 2542
MATERIALS AND METHODS
Animals and treatments
All procedures involving animals were performed with the approval of the Complutense University Animal Experimentation Committee in compliance with the European official regulations. GPR55 KO mice (B6;129S- Gpr55tm1Lex/Mmnc) were generated by Lexicon Pharmaceuticals (The Woodlands, TX, USA) and were obtained through the Mutant Mouse
Regional Resource Centers. This strain has a 129/SvEvBrd x C57BL6/J-mixed genetic background. As pure C57BL6/J mice are refractory to DMBA/ TPA-induced carcinogenesis,9 GPR55 KO animals were bred to the sensitive 129/Ola mice. Heterozygous animals from these progeny were intercrossed to generate the WT and GPR55 KO littermates used in this study. For the two-stage carcinogenesis experiments, tumors were induced on the shaved dorsal skin of WT (n 1⁄4 11) and GPR55 KO (n 1⁄4 11) mice by topical application of a single dose of DMBA (32 mg in 200 ml of acetone), followed by twice-weekly applications of TPA (12.5mg in 200ml of acetone) for 22 weeks. The number of tumors (42mm in diameter) was recorded weekly. TPA treatment was ended after 22 weeks, and animals were left untreated until week 38 after DMBA initiation to allow the development of carcinomas. Tumors were collected at different time points and histologically typed as previously described.12 For the acute TPA experiments, a single dose of TPA (12.5 mg) or the corresponding vehicle (200 ml of acetone) was applied onto the shaved dorsal skin of WT (n 1⁄4 10) and GPR55 KO (n 1⁄4 10) littermates. At the end of the experiments, mice were killed and tumors or skin were collected. Samples were divided into portions that were (1) frozen in Tissue-Tek (Sakura Finetek Europe, Zoeterwoude, The Netherlands) for immunofluorescence staining, (2) fixed in formalin for immunofluorescence, immunohistochemistry and H&E staining, or (3) snap-frozen for protein and RNA extraction. Samples were stored at -80 degrees C until analysis (except formalin-fixed samples, which were kept at room temperature.
Cell cultures, plasmids and transfections
The origin and culture conditions of the PDV-transformed murine keratinocytes has been described elsewhere.12 To stably knockdown GPR55, double-stranded oligonucleotides encoding shRNA against mouse GPR55 were inserted into the psi-U6-EGFP vector (GeneCopoeia, Rockville, MD, USA). A psi-U6-EGFP-unrelated sequence vector was used as control. Transfections were carried out using Lipofectamine-plus (Invitrogen, Barcelona, Spain) according to the manufacturer’s instructions. Cells were selected with puromycin (Sigma, St Louis, MO, USA), and individual clones were isolated using a FACS Vantage sorter (Becton Dickinson, Madrid, Spain). Cell viability was determined in 10% FBS (fetal bovine serum)- containing cell cultures by the MTT test (Sigma) according to manufacturer’s instructions.
Anchorage-independent growth
Cells (5 x103 in 1.5 ml) were suspended in 0.35% agar in DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10% FBS, layered on top of a solid 0.5% agar base in six-well plates, and incubated at 37 1C and 5% CO2 for 30 days. The resulting colonies were morphologically assessed and quantified after staining with crystal violet (0.05% in 25% methanol).
Cell invasion
Cells (2.5 x 104) were suspended in 500 ml DMEM supplemented with 0.1% FBS and loaded onto the upper compartment of BD BioCoat Matrigel Invasion Chambers (BD Biosciences, Bedford, MA, USA). FBS (5%) was used as chemoattractant in the lower compartment. Cell invasion was quantified by staining migrated cell nuclei with DAPI (40,6-diamidino-2-phenylindole) (Roche Applied Science, Barcelona, Spain).
Subcutaneous xenografts
For tumorigenicity assays, 106 viable cells were subcutaneously injected into the flank of 6-week-old athymic mice (Harlan Interfauna Iberica, Barcelona, Spain). Tumors were routinely measured and volume was calculated as (4p/3) x (width/2)2 x (length/2). Fifty days after tumor detection, animals were killed and tumors were collected and processed as described above.
Immunofluoresce analysis
Tissue-Tek or paraffin-embedded sections were fixed in 10% paraformal- dehyde and were subjected to heat-induced antigen retrieval in citrate buffer before exposure to primary antibodies. Anti-GPR55 antibody, the corresponding blocking peptide and anti-proliferating cell nuclear antigen antibody were from Abcam (Cambridge, UK). Anti-CD45R(B220) Alexa Fluor 647 antibody was from BD Biosciences. Secondary anti-rabbit antibodies Alexa Fluor 594 and Alexa Fluor 488 were from Invitrogen. Cell nuclei were stained with DAPI (Invitrogen). Fluorescence confocal images were acquired by using Leica TCS-SP2 software (Leica, Wetzlar, Germany).
Cell invasion
Cells (2.5 x 104) were suspended in 500 ml DMEM supplemented with 0.1% FBS and loaded onto the upper compartment of BD BioCoat Matrigel Invasion Chambers (BD Biosciences, Bedford, MA, USA). FBS (5%) was used as chemoattractant in the lower compartment. Cell invasion was quantified by staining migrated cell nuclei with DAPI (40,6-diamidino-2-phenylindole) (Roche Applied Science, Barcelona, Spain).
Subcutaneous xenografts
For tumorigenicity assays, 106 viable cells were subcutaneously injected into the flank of 6-week-old athymic mice (Harlan Interfauna Iberica, Barcelona, Spain). Tumors were routinely measured and volume was calculated as (4p/3) x (width/2)2 x (length/2). Fifty days after tumor detection, animals were killed and tumors were collected and processed as described above.
Immunofluoresce analysis
Tissue-Tek or paraffin-embedded sections were fixed in 10% paraformal- dehyde and were subjected to heat-induced antigen retrieval in citrate buffer before exposure to primary antibodies. Anti-GPR55 antibody, the corresponding blocking peptide and anti-proliferating cell nuclear antigen antibody were from Abcam (Cambridge, UK). Anti-CD45R(B220) Alexa Fluor 647 antibody was from BD Biosciences. Secondary anti-rabbit antibodies Alexa Fluor 594 and Alexa Fluor 488 were from Invitrogen. Cell nuclei were stained with DAPI (Invitrogen). Fluorescence confocal images were acquired by using Leica TCS-SP2 software (Leica, Wetzlar, Germany).
Immunohistochemical analysis
Paraffin-embedded sections from tumors were deparaffinized and rehydrated using standard protocols and subjected to heat-induced antigen retrieval in citrate buffer. Endogenous peroxidase blockade and unspecific binding reduction were achieved by successive incubation with 3% H2O2 and 5% goat serum (Abcam) supplemented with 3% BSA and 1% Triton X-100 (Sigma), respectively. Samples were then incubated with anti- GPR55 antibody and further processed with appropriate secondary antibodies following the LSAB2 horseradish peroxidase kit’s instructions (Dako cytomation, Carpenteria, CA, USA). Images were acquired with Cell^A software (Olympus, Mu ̈nster, Germany)
Human tumor samples
Two series of skin (18 squamous cell carcinomas and 8 non-tumor skin specimens) and larynx samples (27 carcinomas and 7 non-tumor tissues) were obtained from the CNIO Tumor Bank Core Unit (Madrid, Spain). An additional data set corresponding to 32 oral squamous cell carcinomas plus 6 normal mucosas13 was analyzed. In this case, microarray data were obtained from GEO database, with the accession number GSE10121.
RT– and real-time quantitative–PCR
RNA was isolated with Trizol Reagent (Invitrogen) with the Real Star Kit (Durviz, Valencia, Spain), and cDNA was obtained with Transcriptor Reverse Transcriptase (Roche Applied Science). The primers used for RT– and RTQ (real-time quantitative)–PCR are in Supplementary Table 1. For RTQ–PCR, probes were from the Universal Probe Library (Roche Applied Science) and multispecies 18S RNA was used as reference. For RT–PCR, GAPDH was used as reference.
Western blot analysis
Cell lysates from tumors and cell lines were subjected to SDS–PAGE, and proteins transferred onto PVDF membranes. Blots were incubated with the following antibodies: anti-GPR55 (Abcam), anti-phospho-ERK (Thr202/ Tyr204), anti-ERK, anti-phospho-Akt (Ser473), cyclin D1 (Cell Signaling Technology, Danvers, MA, USA) and anti-a-tubulin (Sigma). Luminograms were obtained with the Amersham Enhanced Chemiluminescence Detection Kit (GE Healthcare, Uppsala, Sweden) and densitometric analysis was performed with Quantity One software (Bio-Rad, Madrid, Spain).
Statistical analysis
ANOVA with a post-hoc analysis by the Student–Newman–Keuls’ test
was routinely used. Unless otherwise stated, data are expressed as mean±s.e.m.
Paraffin-embedded sections from tumors were deparaffinized and rehydrated using standard protocols and subjected to heat-induced antigen retrieval in citrate buffer. Endogenous peroxidase blockade and unspecific binding reduction were achieved by successive incubation with 3% H2O2 and 5% goat serum (Abcam) supplemented with 3% BSA and 1% Triton X-100 (Sigma), respectively. Samples were then incubated with anti- GPR55 antibody and further processed with appropriate secondary antibodies following the LSAB2 horseradish peroxidase kit’s instructions (Dako cytomation, Carpenteria, CA, USA). Images were acquired with Cell^A software (Olympus, Mu ̈nster, Germany)
Human tumor samples
Two series of skin (18 squamous cell carcinomas and 8 non-tumor skin specimens) and larynx samples (27 carcinomas and 7 non-tumor tissues) were obtained from the CNIO Tumor Bank Core Unit (Madrid, Spain). An additional data set corresponding to 32 oral squamous cell carcinomas plus 6 normal mucosas13 was analyzed. In this case, microarray data were obtained from GEO database, with the accession number GSE10121.
RT– and real-time quantitative–PCR
RNA was isolated with Trizol Reagent (Invitrogen) with the Real Star Kit (Durviz, Valencia, Spain), and cDNA was obtained with Transcriptor Reverse Transcriptase (Roche Applied Science). The primers used for RT– and RTQ (real-time quantitative)–PCR are in Supplementary Table 1. For RTQ–PCR, probes were from the Universal Probe Library (Roche Applied Science) and multispecies 18S RNA was used as reference. For RT–PCR, GAPDH was used as reference.
Western blot analysis
Cell lysates from tumors and cell lines were subjected to SDS–PAGE, and proteins transferred onto PVDF membranes. Blots were incubated with the following antibodies: anti-GPR55 (Abcam), anti-phospho-ERK (Thr202/ Tyr204), anti-ERK, anti-phospho-Akt (Ser473), cyclin D1 (Cell Signaling Technology, Danvers, MA, USA) and anti-a-tubulin (Sigma). Luminograms were obtained with the Amersham Enhanced Chemiluminescence Detection Kit (GE Healthcare, Uppsala, Sweden) and densitometric analysis was performed with Quantity One software (Bio-Rad, Madrid, Spain).
Statistical analysis
ANOVA with a post-hoc analysis by the Student–Newman–Keuls’ test
was routinely used. Unless otherwise stated, data are expressed as mean±s.e.m.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We are indebted to the members of our laboratories and to Dr MM Caffarel for critical discussion on this work. We thank Dr Manuel M Morente, Head of the CNIO Tumor Bank Core Unit, for kindly provided us with human tumor samples. This work was supported by grants from Fondo de Investigaciones Sanitarias (PI080253 to CS), Fundacio ́ n Mutua Madrilen ̃ a (to CS), GW Pharmaceuticals/Otsuka Pharmaceuticals (to CS), Comunidad de Madrid (S2010/BMD-2038 to MG) and Spanish Ministry of Science and Innovation (SAF2010-19152 to MQ and SAF2008-00121 and SAF2011-26122-C02- 01 to JMP). EPG and CA were the recipients of a Postdoctoral Research Contract from Fundacio ́n Cient ́ıfica Asociacio ́n Espan ̃ola Contra el Ca ́ncer (AECC) and a PFIS PhD studentship from the Spanish Ministry of Science and Innovation, respectively.

The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We are indebted to the members of our laboratories and to Dr MM Caffarel for critical discussion on this work. We thank Dr Manuel M Morente, Head of the CNIO Tumor Bank Core Unit, for kindly provided us with human tumor samples. This work was supported by grants from Fondo de Investigaciones Sanitarias (PI080253 to CS), Fundacio ́ n Mutua Madrilen ̃ a (to CS), GW Pharmaceuticals/Otsuka Pharmaceuticals (to CS), Comunidad de Madrid (S2010/BMD-2038 to MG) and Spanish Ministry of Science and Innovation (SAF2010-19152 to MQ and SAF2008-00121 and SAF2011-26122-C02- 01 to JMP). EPG and CA were the recipients of a Postdoctoral Research Contract from Fundacio ́n Cient ́ıfica Asociacio ́n Espan ̃ola Contra el Ca ́ncer (AECC) and a PFIS PhD studentship from the Spanish Ministry of Science and Innovation, respectively.
